

IPC分类号 : C09K11/66I,B82Y20/00I,B82Y40/00I,C01G21/00I








专利摘要
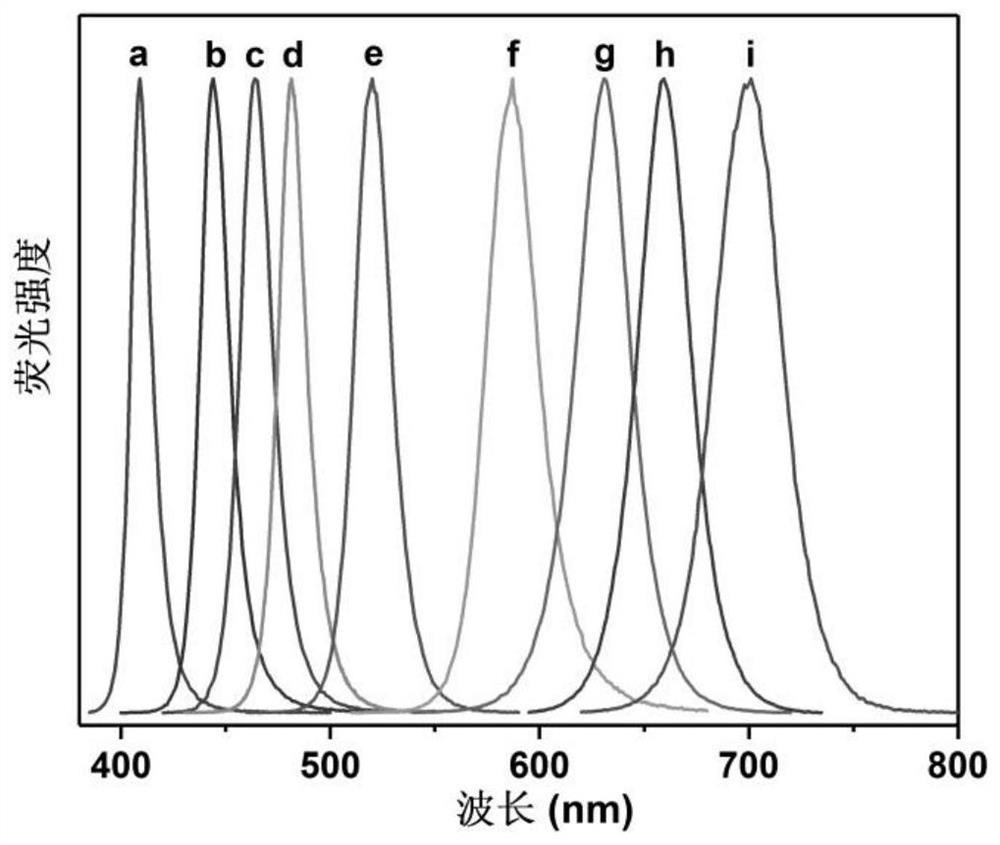
本发明公开了一种ABX3型全无机钙钛矿纳米晶的制备方法。所述制备过程中溶剂用量少、原料单价较低(如以价格便宜的醋酸铅等替代价格昂贵的PbX2原料),制备过程中对环境的要求不高,无需使用手套箱,操作程序简单,大大降低了原料和仪器的成本。所述纳米晶在制备过程中可在0至300℃的宽温度区间合成目标产物,通过改变原料配比、溶剂配比、反应温度和/或反应时间可以控制纳米晶的形貌、物相、尺寸和发光性能,例如在大于等于0℃且小于90℃可制备出钙钛矿纳米线,在90‑300℃可制备出钙钛矿量子点。所述纳米晶稳定性好、荧光量子产率高(26%‑80%)、可实现全可见谱段(400‑700nm)发光,所述制备方法工艺简单、耗时短、成本低、易于放大合成,适合工业化大规模制备。
权利要求
1.一种ABX
(1)将含有A的化合物、含有B的化合物,在惰性气氛中溶解于表面活性剂中,或溶解于含有表面活性剂的溶剂中,得到混合溶液;
(2)向步骤(1)的混合溶液中加入含有X的化合物,反应,获得沉淀物,即制备得到ABX
步骤(1)中,所述含有A的化合物选自A的碳酸盐、氧化物、氢氧化物、硝酸盐、硫酸盐、草酸盐、硼酸盐、钒酸盐、钨酸盐、钼酸盐或铬酸盐中的一种或几种的组合;
所述含有B的化合物选自B的醋酸盐、碳酸盐、氧化物、氢氧化物、硝酸盐、硫酸盐、草酸盐、硼酸盐、钒酸盐、钨酸盐、钼酸盐或铬酸盐中的一种或几种的组合;
步骤(1)中,溶解温度为120-200℃;
步骤(1)中,所述表面活性剂选自油酸、油胺和三正辛基膦中的一种或几种的组合;所述溶剂选自十八烯;
步骤(2)中,金属离子(A+B)与X的摩尔比为1:(0.1-3);
所述含有X的化合物选自卤化氢HX;
所述纳米晶选自纳米线,且步骤(2)中,在大于等于0℃且小于90℃的温度范围内制备得到钙钛矿纳米线。
2.根据权利要求1所述的制备方法,其特征在于,所述方法还包括如下步骤:
(3)将步骤(2)的沉淀物冷却、离心、洗涤,将产物分散在非极性有机溶剂中,得到ABX
3.根据权利要求1所述的制备方法,其特征在于,所述纳米线的至少一维尺寸在2-100nm范围内。
4.根据权利要求1所述的制备方法,其特征在于,所述X选自F、Cl或Br中的一种或几种的组合,或者,所述X选自F、Br或I中的一种或几种的组合。
5.根据权利要求4所述的制备方法,其特征在于,所述X选自Cl和/或Br,或者,为Br和/或I。
6.根据权利要求1所述的制备方法,其特征在于,步骤(1)中,所述惰性气氛为氮气和/或氩气。
7.根据权利要求1所述的制备方法,其特征在于,步骤(1)中,溶解的时间为5分钟-72小时。
8.根据权利要求1所述的制备方法,其特征在于,油酸、油胺、三正辛基膦和十八烯的摩尔比为1:(0.5-5):(0-5):(0-10)。
9.根据权利要求1所述的制备方法,其特征在于,步骤(1)中,A和B的摩尔比为(0.5-2):3,金属离子(A+B)与表面活性剂的摩尔比为1:(5-100)。
10.根据权利要求1所述的制备方法,其特征在于,步骤(2)中,所述反应的温度为20-80°C。
11.根据权利要求2所述的制备方法,其特征在于,步骤(3)中,所述沉淀物的冷却速度为(0.5-30)°C/秒。
12.根据权利要求1所述的制备方法,其特征在于,步骤(3)中,所述洗涤使用丙酮、乙腈、正丁醇、异丙醇、叔丁醇、乙醚、甲基乙基甲酮、辛烷、环己烷或甲苯中的一种或几种的组合进行洗涤。
13.根据权利要求1-12任一项所述的制备方法,其特征在于,所述ABX
说明书
技术领域
本发明属于纳米晶发光材料技术领域,具体涉及一种ABX3型全无机钙钛矿纳米晶的制备方法。
背景技术
全无机钙钛矿纳米晶(CsPbX3,X=Cl,Br,I)具有吸收截面大、荧光量子产率高、可调谐荧光发射波长等优异的光学性能,在太阳能电池、发光二极管以及激光等光电器件中展现出了极大的应用前景。目前,制备全无机钙钛矿纳米晶主要有高温注射法与常温过饱和重结晶两种方法。高温注射法是分别将 PbX2和CsCO3两种原料加热溶解于油酸、油胺和十八烯的混合溶剂中,形成Pb 油酸配合物溶液和Cs油酸配合物溶液,然后将Cs油酸配合物溶液在高温下注射到Pb油酸配合物溶液中反应一段时间后冰浴淬冷,离心洗涤后获得CsPbX3钙钛矿纳米晶(参考文献:M.V.Kovalenko et al.,Nano Lett.2015,15,3692)。这种方法获得的钙钛矿纳米晶质量较高,但是CsCO3在溶解降温过程中容易析出,需要保持高于100℃进行注射,且溶剂用量大;另外,PbX2特别是PbBr2、PbI2价格昂贵,使得材料制备成本高,不适合用于工业化生产。常温过饱和重结晶的方法是将PbX2和CsX溶解在DMF和/或DMSO等有机溶剂中,并加入少量油酸和油胺作为表面活性剂,然后将含Pb、Cs和X离子的DMF和/或DMSO溶液逐滴加入甲苯中,由于这些离子在甲苯中溶解度非常低,Pb、Cs和X离子可以快速反应并结晶析出CsPbX3钙钛矿纳米晶(参考文献:H.B.Zeng et al.,Adv.Funct.Mater.2016,26,2435)。这种方法的优点是可以在常温下进行反应,但PbX2和 CsX在DMF和/或DMSO中的溶解度较低,需要大量溶剂来溶解,且PbX2和CsX 价格高,因此也不适用于工业化生产。虽然人们在这两种方法的基础进行了改进合成,比如微波辅助合成、超声辅助合成等,但使用的Pb源都是PbX2,而且溶剂用量大、成本高。发展低成本且适用于工业化放大生产的钙钛矿纳米晶合成方法对开发该类材料至关重要,也是其获得实际应用的前提。
发明内容
为了改善现有技术的不足,本发明的目的是提供一种ABX3型全无机钙钛矿纳米晶的制备方法,所述方法溶剂用量少、且无需使用价格昂贵的PbX2等原料,大大降低了原料成本,所述制备过程可在0至300℃的宽温度区间合成目标产物,通过改变原料配比、溶剂配比、反应温度和/或反应时间等条件可以控制纳米晶的形貌(如制备得到量子点或纳米线)、物相(如立方晶系)、尺寸(量子点的粒径为8-20nm;纳米线的至少一维尺寸在1-100nm范围内)和发光性能,所述制备方法工艺简单、耗时短、成本低、易于放大合成,制备得到的纳米晶稳定性好、荧光量子产率高,适合工业化大规模制备。
本发明是通过如下技术方案实现:
一种ABX3型全无机钙钛矿纳米晶的制备方法,所述制备方法包括如下步骤:
(1)将含有A的化合物,含有B的化合物,含有A和B的化合物,A的单质或B的单质中的至少一种且同时含有A和B元素,在惰性气氛中溶解于表面活性剂中,或溶解于含有表面活性剂的溶剂中,得到混合溶液;
(2)向步骤(1)的混合溶液中加入含有X的化合物或单质,反应,获得沉淀物,即制备得到ABX3型全无机钙钛矿纳米晶,其中,A选自Li,Na,K,Rb 或Cs中的一种或几种的组合,B选自Pb,Sn,Cd,Zn,Ge,Mn,Ni,Mg, Ca,Sr,Ba,Ga,Bi,Cr或Eu中的一种或几种的组合,X选自F,Cl,Br或I 中的一种或几种的组合。
根据本发明,所述方法还包括如下步骤:
(3)将步骤(2)的沉淀物冷却、离心、洗涤,将产物分散在非极性有机溶剂中,得到ABX3型全无机钙钛矿纳米晶溶液;或者,将产物干燥,得到ABX3型全无机钙钛矿纳米晶固体(如固体粉末)。
优选地,所述纳米晶选自量子点或纳米线。
根据本发明,所述量子点为球形或近球形;所述量子点的粒径为8-20nm,优选为10-15nm。
根据本发明,所述纳米线的至少一维尺寸在1-100nm范围内;优选地,所述纳米线的至少一维尺寸在2-50nm范围内。
优选地,所述A选自Cs和/或Rb;还优选为Cs。
优选地,所述B选自Pb,Sn,Cd,Mn,Ni或Zn中的一种或几种的组合,或者,所述B选自Pb,Ca,Sr或Ba中的一种或几种的组合;还优选为Pb和/或Sn,或者,Pb和/或Mn;进一步优选为Pb。
优选地,所述X选自F,Cl或Br中的一种或几种的组合,或者,所述X选自F, Br或I中的一种或几种的组合;还优选为Cl和/或Br,或者,还优选为Br和/或I。
根据本发明,步骤(1)中,所述含有A的化合物、含有B的化合物、含有A 和B的化合物、A的单质或B的单质中的至少一种且同时含有A和B元素选自含有 A和B的化合物,以及任选地含有A的化合物、含有B的化合物、A的单质或B的单质中的至少一种;或者选自含有A的化合物和含有B的化合物,以及任选地含有A和B的化合物、A的单质或B的单质中的至少一种;或者选自含有A的化合物和B的单质,以及任选地含有B的化合物、含有A和B的化合物或A的单质中的至少一种;或者选自含有B的化合物和A的单质,以及任选地含有A的化合物、含有A和B的化合物或B的单质中的至少一种;或者选自A的单质和B的单质,以及任选地含有A和B的化合物、含有A的化合物或含有B的化合物中的至少一种。
根据本发明,步骤(1)中,所述含有A的化合物选自A的碳酸盐,醋酸盐,油酸盐,硬脂酸盐,氧化物,氢氧化物,硝酸盐,硫酸盐,草酸盐,硼酸盐,钒酸盐,钨酸盐,钼酸盐或铬酸盐中的一种或几种的组合,优选为A的碳酸盐和 /或醋酸盐。
根据本发明,步骤(1)中,所述含有B的化合物选自B的醋酸盐,碳酸盐,油酸盐,硬脂酸盐,氧化物,氢氧化物,硝酸盐,硫酸盐,草酸盐,硼酸盐,钒酸盐,钨酸盐,钼酸盐或铬酸盐中的一种或几种的组合,优选为B的醋酸盐和 /或草酸盐。
根据本发明,步骤(1)中,所述含有A和B的化合物选自Cs2PbO2,Ru2PbO2, Cs2Sn2O3,Ru2SnO2,Ru2Sn2O3,CsCdF3或RuCdF3中的一种或几种的组合。
根据本发明,步骤(1)中,所述A的单质选自Li,Na,K,Rb或Cs中的一种或几种的组合。
根据本发明,步骤(1)中,所述B的单质选自Pb,Sn,Cd,Zn,Ge,Mn, Mg,Ca,Sr,Ba,Ga,Bi,Cr或Eu中的一种或几种的组合。
根据本发明,步骤(1)中,所述惰性气氛为氮气和/或氩气。
根据本发明,步骤(1)中,溶解的温度为80-250℃,优选为120-200℃。
根据本发明,步骤(1)中,溶解的时间为5分钟-72小时,优选为10-120分钟。
根据本发明,步骤(1)中,所述表面活性剂选自油酸,油胺,三正辛基膦,三正辛基氧膦,硬脂酸,十二烷基苯磺酸钠,十六烷基三甲基溴化铵,聚丙烯酸,月桂酸,柠檬酸,乙二胺四乙酸或乙二胺四乙酸钠中的一种或几种的组合,优选为油酸,油胺和三正辛基膦中的一种或几种的组合;还优选为油酸和/或油胺。
根据本发明,步骤(1)中,所述溶剂选自十八烯,三辛胺,硬脂酸丁酯,三戊苯,四戊苯,环己烷,正己烷,氯仿或甲苯中的一种或几种的组合,优选为十八烯和/或三辛胺,还优选为十八烯。
根据本发明,步骤(1)中,所述表面活性剂和溶剂的摩尔比为1:(0-200);优选为1:(0-20);采用上述范围的表面活性剂与溶剂的摩尔配比,更有利于含有A和B元素的单质或化合物在其中的溶解,实现在少量溶剂的使用前提下获得更高的钙钛矿纳米晶产率。
优选地,所述油酸,油胺,三正辛基膦和十八烯的摩尔比为1:(0.1-20): (0-20):(0-200),还优选为1:(0.25-10):(0-10):(0-20),进一步优选为 1:(0.5-5):(0-5):(0-10);本领域技术人员应当理解,如果上述某组分不存在,其他组分之间的比例仍适用上述摩尔比。
根据本发明,步骤(1)中,A和B的摩尔比为(0.001-1):1,还优选为(0.5-2): 3;其中,所述的A为以离子形式存在的A离子,示例性地如Li离子,Na离子, K离子,Rb离子或Cs离子中的至少一种;所述的B为以离子形式存在的B离子,示例性地如Pb离子,Sn离子,Cd离子,Zn离子,Ge离子,Mn离子,Ni离子, Mg离子,Ca离子,Sr离子,Ba离子,Ga离子,Bi离子,Cr离子或Eu离子中的至少一种;采用上述范围的A和B的摩尔配比,可以获得不同尺寸、形貌及发光性能的ABX3钙钛矿纳米晶;若其他条件不变时,A:B的摩尔比越小(小于0.001),所得钙钛矿纳米晶的尺寸也越小,发光蓝移,合成产率越低,纳米晶的发光性能较差;A:B的摩尔比大于1时,所得产物将不再是ABX3型钙钛矿纳米晶,其发光性能也急剧下降。
根据本发明,步骤(1)中,金属离子(A+B)与表面活性剂的摩尔比为1: (1-1000),优选为1:(5-100);其中,所述的金属离子(A+B)是指A离子和B离子,金属离子(A+B)的摩尔数是指A离子的摩尔数和B离子的摩尔数之和;采用不同金属离子(A+B)与表面活性剂的摩尔比,可以获得不同尺寸、形貌及发光性能的ABX3型钙钛矿纳米晶;表面活性剂可以减缓金属离子的扩散,若其他条件不变,金属离子(A+B)与表面活性剂的摩尔比越小,钙钛矿纳米晶的成核与生长速率越慢,所得产物的尺寸越小,发光蓝移。
根据本发明,步骤(2)中,所述含有X的单质选自Cl2、Br2或I2。所述含有 X的化合物选自卤化氢HX,AX,BX,MX或LnX3中的一种或几种的组合,其中,A和B的定义如上所述,M为碱土金属离子(如铍(Be)、镁(Mg)、钙(Ca)、锶(Sr)、钡(Ba)、镭(Ra)),Ln为稀土金属离子(如钪(Sc)、钇(Y)、镧(La)、铈(Ce)、镨(Pr)、钕(Nd)、钷(Pm)、钐(Sm)、铕(Eu)、钆(Gd)、铽(Tb)、镝(Dy)、钬(Ho)、铒(Er)、铥(Tm)、镱(Yb)、镥(Lu));优选为卤化氢 HX;通过改变X元素及其配比,可以获得不同组分和发光性能的ABX3型钙钛矿纳米晶;比如通过改变X中Cl、Br或I的比例,所得钙钛矿纳米晶的组分可以从 CsPbCl3,CsPbBr3到CsPbI3演变,其对应的带隙和发射光谱可从400-750nm范围内进行调控,制备得到性能更加优异的产品。
根据本发明,步骤(2)中,所述反应的温度为0-300℃,优选为大于等于0℃且小于90℃,或优选为90-300℃,还优选为大于等于20℃且小于90℃,或优选为90-200℃,还更优选为20-80℃,或优选为150-200℃;采用不同的反应温度,可以获得不同尺寸、形貌及发光性能的ABX3钙钛矿纳米晶;若其他条件不变,反应温度越高,所得钙钛矿纳米晶尺寸越大,发光红移、强度降低;当温度在 90-300℃范围改变时,可以获得不同尺寸和发光性能的钙钛矿量子点;当温度在大于等于0℃且小于90℃范围改变时,通过调控溶剂配比和反应时间,可以获得不同尺寸和发光性能的钙钛矿纳米线,其发光呈双峰发射。
根据本发明,步骤(2)中,所述反应的时间为大于0秒且小于等于72小时,优选为5秒-1小时,还优选为5秒-30分钟;采用不同反应时间,可以获得不同尺寸、形貌及发光性能的ABX3钙钛矿纳米晶;在选定的时间范围内,若其他条件不变,反应时间越长,所得钙钛矿纳米晶尺寸越大,其发光红移、强度降低。
根据本发明,步骤(2)中,金属离子(A+B)与X的摩尔比为1:(0.01-10),优选为1:(0.1-5),还优选为1:(0.1-3);若X选用AX和/或BX时,此处的金属离子(A+B)为步骤(1)中的金属离子A和金属离子B,以及步骤(2)中的金属离子A和/或金属离子B;若X选用非AX和/或BX时,此处的金属离子(A+B) 仅为步骤(1)中的金属离子A和金属离子B;采用不同金属离子(A+B)与X的摩尔比,可以获得不同尺寸、形貌及发光性能的ABX3钙钛矿纳米晶;若其他条件不变,(A+B)与X的摩尔比越小,所得钙钛矿纳米晶的尺寸也越小,其发光蓝移、强度增加。
根据本发明,步骤(3)中,所述沉淀物的冷却速度为(0.01-200)℃/秒,优选为(0.5-30)℃/秒;所述沉淀物的冷却可以通过使用常规的水浴、冰水浴和/ 或冰浴实现,当然也可以通过其他的降温仪器实现对沉淀物的冷却处理,通过控制沉淀物的冷却速度,可以获得不同尺寸、形貌及发光性能的ABX3钙钛矿纳米晶;冷却速度越快,所得钙钛矿纳米晶尺寸越小,其发光蓝移、强度增加。
根据本发明,步骤(3)中,所述离心是将获得的沉淀物与反应体系分开,通过离心分离的方式获得固体产物;所述洗涤是去除残余在固体产物表面的表面活性剂;所述洗涤的方式可以是过滤洗涤或是离心洗涤;所述洗涤可以使用有机溶剂如丙酮,乙腈,正丁醇,异丙醇,叔丁醇,乙醚,甲基乙基甲酮,辛烷,环己烷或甲苯中的一种或几种的组合进行洗涤,优选丙酮和/或环己烷。
根据本发明,步骤(3)中,将洗涤后的产物干燥获得钙钛矿纳米晶固体粉末;所述干燥的温度为30℃-100℃,优选为50℃-80℃。
根据本发明,步骤(3)中,将洗涤后的产物分散在非极性有机溶剂中获得钙钛矿纳米晶溶液;所述非极性有机溶剂分散剂选自正己烷,环己烷,三氯甲烷,二氯甲烷或甲苯中的一种或几种的组合,优选为环己烷和/或甲苯。
优选地,所述ABX3选自CsPbX3,所述CsPbX3选自CsPb(Clx/Bry),其中 x+y=3,0≤x≤3,0≤y≤3;或者,所述CsPbX3选自CsPb(Brx/Iy),其中x+ y=3,0≤x≤3,0≤y≤3。
优选地,所述ABX3选自APbX3,所述APbX3选自(Csx/Ruy)PbX3,其中x+ y=1,0≤x≤1,0≤y≤1。
优选地,所述ABX3选自CsBX3,所述CsBX3选自Cs(Pbx/Mny)X3,其中x+ y=1,0≤x≤1,0≤y≤1。
本发明的有益效果在于:
1.本发明的ABX3型全无机钙钛矿纳米晶在制备过程中溶剂用量少、原料单价较低(如以价格便宜的醋酸铅等替代价格昂贵的PbX2原料),制备过程中对环境的要求不高,无需使用手套箱,操作程序简单,大大降低了原料和仪器的成本。
2.本发明的ABX3型全无机钙钛矿纳米晶在制备过程中是直接将A和B的原料混合溶解,避免因两种原料分开溶解而存在A原料如CsCO3降温过程易析出的风险,导致的需将A原料重新加热至100℃以上进行注射反应的缺点。
3.本发明的ABX3型全无机钙钛矿纳米晶在制备过程中使用的含有X的化合物或单质,如卤化氢HX作为X源进行注射反应,避免了价格昂贵的PbX2原料的使用,通过改变卤素X的注射种类可以改变产物的组分,通过改变HX的注射量可以改变产物的尺寸、形貌和物相,从而获得不同光学性能的ABX3钙钛矿纳米晶,如纳米线或量子点。
4.本发明的ABX3型全无机钙钛矿纳米晶在制备过程中可在0至300℃的宽温度区间合成目标产物,通过改变原料配比、溶剂配比、反应温度和/或反应时间可以控制纳米晶的形貌、物相、尺寸和发光性能,例如在大于等于0℃且小于90℃可制备出钙钛矿纳米线,在90-300℃可制备出钙钛矿量子点。
5.本发明的ABX3型全无机钙钛矿纳米晶稳定性好、荧光量子产率高 (26%-80%)、可实现全可见谱段(400-700nm)发光,所述制备方法工艺简单、耗时短、成本低、易于放大合成,适合工业化大规模制备。
附图说明
图1中的a-i)分别对应实施例1-9中CsPbCl3,CsPbCl2Br,CsPbCl1.5Br1.5,CsPbClBr2,CsPbBr3,CsPbBr2I,CsPbBr1.5I1.5,CsPbBrI2,CsPbI3钙钛矿量子点的X射线粉末衍射图。
图2中的a1-e4)分别对应实施例1,3,5,7,9中a)CsPbCl3,b)CsPbCl1.5Br1.5, c)CsPbBr3,d)CsPbBr1.5I1.5,e)CsPbI3钙钛矿量子点的1)透射电镜图,2) 粒径统计分布图,3)选区电子衍射图和4)EDS(能量色散)能谱图。
图3中的a1-a9)分别对应实施例1-9中CsPbCl3,CsPbCl2Br,CsPbCl1.5Br1.5,CsPbClBr2,CsPbBr3,CsPbBr2I,CsPbBr1.5I1.5,CsPbBrI2,CsPbI3钙钛矿量子点的环己烷溶液(钙钛矿量子点的浓度为1mg/mL)日光下的发光照片; b1-b9)分别对应其在365nm紫外灯照射下的发光照片。
图4中的a-i)分别对应实施例1-9中CsPbCl3,CsPbCl2Br,CsPbCl1.5Br1.5,CsPbClBr2,CsPbBr3,CsPbBr2I,CsPbBr1.5I1.5,CsPbBrI2,CsPbI3钙钛矿量子点的吸收光谱。
图5中的a-i)分别对应实施例1-9中CsPbCl3,CsPbCl2Br,CsPbCl1.5Br1.5,CsPbClBr2,CsPbBr3,CsPbBr2I,CsPbBr1.5I1.5,CsPbBrI2,CsPbI3钙钛矿量子点的荧光发射光谱,激发波长为365nm。
图6中的a-i)分别对应实施例1-9中CsPbCl3,CsPbCl2Br,CsPbCl1.5Br1.5,CsPbClBr2,CsPbBr3,CsPbBr2I,CsPbBr1.5I1.5,CsPbBrI2,CsPbI3钙钛矿量子点的荧光衰减曲线。
图7中的a-e)分别对应实施例10中CsPbBr3钙钛矿纳米线的a)透射电镜图片,b)高分辨透射电镜图片,c)选区电子衍射图,d)X射线粉末衍射图,e) 荧光发射光谱,激发波长为365nm。
图8中的a-e)分别对应实施例11中CsPbCl3:Mn
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的保护范围。此外,应理解,在阅读了本发明所公开的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本发明所限定的保护范围之内。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法;下述实施例中所用的试剂、材料等,如无特殊说明,均可从商业途径得到。
仪器和设备:
本发明实施例产品进行粉末衍射表征使用的仪器型号为MiniFlex2,厂家为Rigaku,铜靶辐射波长为λ=0.154187nm。
本发明实施例产品进行X射线能谱分析使用的仪器型号为JEM-2010,厂家为JEOL。
本发明实施例产品进行透射电镜检测使用的仪器型号为TECNAI G
本发明实施例产品进行紫外可见吸收光谱表征使用的仪器型号为 Lambda365,厂家为Perkin-Elmer。
本发明实施例产品进行荧光发射光谱、荧光寿命表征使用的仪器型号为 FLS980,厂家为Edinburgh,激发光源为氙灯和390nm LD脉冲激光器。
实施例1:CsPbCl3钙钛矿量子点的制备。
称取0.5mmol醋酸铅、0.1mmol碳酸铯,然后加入1mL油酸、1mL油胺、 1mL三正辛基膦和10mL十八烯,通氮气加热至120℃并保温10分钟,形成透明溶液A,然后升温至180℃;向溶液A中快速注射124μL盐酸,保温10秒,用冰浴快速冷却(冷却速度约为10-15℃/秒)至室温;先离心分离再用10mL环己烷和10mL丙酮洗涤1次,并将沉淀物分散在30mL环己烷中,得到粒径为11.0 nm左右的CsPbCl3钙钛矿量子点。
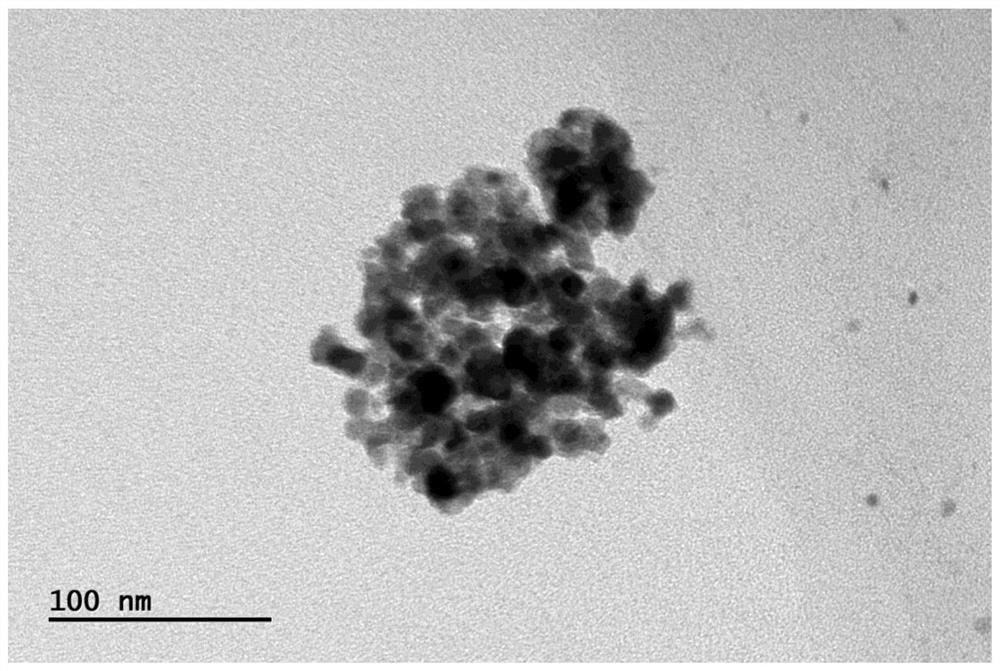
X射线粉末衍射图(图1a)表明该量子点具有良好的结晶性,其衍射峰位置和相对强度与立方相CsPbCl3的PDF标准卡片(JCPDS NO.75-0411)一致,属于立方晶系;透射电镜和粒径分布图(图2a1和2a2)表明该量子点分散性好、形貌均一,粒径约为11.0nm;选区电子衍射图片(图2a3)表明该量子点具有良好的晶化;EDS能谱图(图2a4)指明了该量子点存在Cs、Pb、Cl元素;该量子点的环己烷溶液(钙钛矿量子点的浓度为1mg/mL)在日光下的照片(图3a1) 呈透明颜色,在365nm紫外灯下表现出强紫光发射(图3b1);吸收光谱(图4a) 表明该量子点在紫外区域具有强吸收,吸收边约为410nm;365nm激发下的荧光发射光谱(图5a)表明该量子点在410nm具有强发光,半峰宽约为12nm;荧光衰减曲线(图6a)表明该量子点的有效荧光寿命约为1.8ns;荧光量子产率测试表明该量子点的绝对荧光量子产率约为26%。
实施例2:CsPbCl2Br钙钛矿量子点的制备。
称取0.5mmol草酸铅、0.2mmol醋酸铯,然后加入1mL油酸、1mL油胺、 1.5mL三正辛基膦和20mL十八烯,通氮气加热至120℃并保温20分钟,形成透明溶液A,然后升温至200℃;向溶液A中快速注射83μL盐酸和58μL溴化氢,保温5秒,自然冷却至室温;先离心分离再用10mL环己烷和10mL丙酮洗涤1次,并将沉淀物分散在30mL环己烷中,得到粒径为15.0nm左右的CsPbCl2Br钙钛矿量子点。
X射线粉末衍射图(图1b)表明该量子点具有良好的结晶性,其衍射峰位置和相对强度与立方相CsPbCl3和CsPbBr3的PDF标准卡片(JCPDS NO.75-0411 和75-0412)基本一致,介于两者之间,属于立方晶系;该量子点的环己烷溶液 (钙钛矿量子点的浓度为1mg/mL)在日光下的照片(图3a2)呈紫色,在365nm 紫外灯下表现出强蓝紫光发射(图3b2);吸收光谱(图4b)表明该量子点在紫外和紫光区域具有强吸收,吸收边约为425nm;365nm激发下的荧光发射光谱 (图5b)表明该量子点在444nm具有强发光,半峰宽约为16nm;荧光衰减曲线(图6b)表明该量子点的有效荧光寿命约为3.8ns;荧光量子产率测试表明该量子点的绝对荧光量子产率约为36%。
实施例3:CsPbCl1.5Br1.5钙钛矿量子点的制备。
称取0.5mmol醋酸铅、0.05mmol碳酸铯,然后加入0.5mL油酸、0.5mL 油胺和6mL十八烯,通氮气加热至120℃并保温30分钟,形成透明溶液A,然后升温至190℃;向溶液A中快速注射62μL盐酸和87μL溴化氢,保温5秒,用冰水浴快速冷却至室温;先离心分离再用5mL环己烷和5mL丙酮洗涤1次,并将沉淀物分散在30mL环己烷中,得到粒径为13.0nm左右的CsPbCl1.5Br1.5钙钛矿量子点。
X射线粉末衍射图(图1c)表明该量子点具有良好的结晶性,其衍射峰位置和相对强度与立方相CsPbCl3和CsPbBr3的PDF标准卡片(JCPDS NO.75-0411 和75-0412)基本一致,介于两者之间,属于立方晶系;透射电镜和粒径分布图 (图2b1和2b2)表明该量子点分散性好、形貌均一,粒径约为13.0nm;选区电子衍射图片(图2b3)表明该量子点具有良好的晶化;EDS能谱图(图2b4)指明了该量子点存在Cs、Pb、Cl、Br元素;该量子点的环己烷溶液(钙钛矿量子点的浓度为1mg/mL)在日光下的照片(图3a3)呈蓝色,在365nm紫外灯下表现出强蓝光发射(图3b3);吸收光谱(图4c)表明该量子点在紫外到蓝光区域具有强吸收,吸收边约为460nm;365nm激发下的荧光发射光谱(图5c)表明该量子点在465nm具有强发光,半峰宽约为17nm;荧光衰减曲线(图6c)表明该量子点的有效荧光寿命约为8.9ns;荧光量子产率测试表明该量子点的绝对荧光量子产率约为62%。
实施例4:CsPbClBr2钙钛矿纳米晶的制备。
称取1mmol醋酸铅、0.2mmol碳酸铯,然后加入3mL油酸和3mL油胺,通氮气加热至120℃并保温30分钟,形成透明溶液A,然后升温至170℃;向溶液A中快速注射84μL盐酸和232μL溴化氢,保温20秒,用水浴快速冷却(冷却速度约为0.5-4℃/秒)至室温;加入5mL环己烷和5mL丙酮离心分离,再用10 mL环己烷和10mL丙酮洗涤1次,并将沉淀物分散在50mL环己烷中,得到粒径为16.0nm左右的CsPbClBr2钙钛矿量子点。
X射线粉末衍射图(图1d)表明该量子点具有良好的结晶性,其衍射峰位置和相对强度与立方相CsPbCl3和CsPbBr3的PDF标准卡片(JCPDS NO.75-0411 和75-0412)基本一致,介于两者之间,属于立方晶系;该量子点的环己烷溶液 (钙钛矿量子点的浓度为1mg/mL)在日光下的照片(图3a4)呈蓝绿色,在365 nm紫外灯下表现出强蓝绿光发射(图3b4);吸收光谱(图4d)表明该量子点在紫外到蓝光区域具有强吸收,吸收边约为480nm;365nm激发下的荧光发射光谱(图5d)表明该量子点在481nm具有强发光,半峰宽约为18nm;荧光衰减曲线(图6d)表明该量子点的有效荧光寿命约为8.2ns;荧光量子产率测试表明该量子点的绝对荧光量子产率约为61%。
实施例5:CsPbBr3钙钛矿量子点的制备。
称取0.5mmol醋酸铅、0.08mmol碳酸铯,然后加入1.5mL油酸、1.5mL 油胺和8mL十八烯,通氮气加热至120℃并保温30分钟,形成透明溶液A,然后升温至170℃;向溶液A中快速注射173μL溴化氢,保温10秒,用冰浴快速冷却(冷却速度约为10-15℃/秒)至室温;先离心分离再用5mL环己烷和5mL丙酮洗涤1次,并将沉淀物分散在30mL环己烷中,得到粒径为13.3nm左右的 CsPbBr3钙钛矿量子点。
X射线粉末衍射图(图1e)表明该量子点具有良好的结晶性,其衍射峰位置和相对强度与立方相CsPbBr3的PDF标准卡片(JCPDS NO.75-0412)一致,属于立方晶系;透射电镜和粒径分布图(图2c1和2c2)表明该量子点分散性好、形貌均一,粒径约为13.3nm;选区电子衍射图片(图2c3)表明该量子点具有良好的晶化;EDS能谱图(图2c4)指明了该量子点存在Cs、Pb、Br元素;该量子点的环己烷溶液(钙钛矿量子点的浓度为1mg/mL)在日光下的照片(图3a5) 呈绿色,在365nm紫外灯下表现出强绿光发射(图3b5);吸收光谱(图4e)表明该量子点在紫外到绿光区域具有强吸收,吸收边约为520nm;365nm激发下的荧光发射光谱(图5e)表明该量子点在520nm具有强发光,半峰宽约为20nm;荧光衰减曲线(图6e)表明该量子点的有效荧光寿命约为21ns;荧光量子产率测试表明该量子点的绝对荧光量子产率约为80%。
实施例6:CsPbBr2I钙钛矿量子点的制备。
称取0.5mmol醋酸铅、0.2mmol醋酸铯,然后加入1.5mL油酸、1.5mL 油胺和10mL十八烯,通氮气加热至120℃并保温40分钟,形成透明溶液A,然后升温至160℃;向溶液A中快速注射116μL溴化氢和66μL碘化氢,保温10秒,用冰浴快速冷却至室温;先离心分离再用1mL环己烷洗涤1次,并将沉淀物分散在30mL环己烷中,得到粒径为12.0nm左右的CsPbBr2I钙钛矿量子点。
X射线粉末衍射图(图1f)表明该量子点具有良好的结晶性,其衍射峰位置和相对强度与立方相CsPbBr3的PDF标准卡片(JCPDS NO.75-0412)基本一致,属于立方晶系;该量子点的环己烷溶液(钙钛矿量子点的浓度为1mg/mL)在日光下的照片(图3a6)呈橙色,在365nm紫外灯下表现出强橙黄光发射(图3b6);吸收光谱(图4f)表明该量子点在紫外到黄光区域具有强吸收,吸收边约为590 nm;365nm激发下的荧光发射光谱(图5f)表明该量子点在587nm具有强发光,半峰宽约为28nm;荧光衰减曲线(图6f)表明该量子点的有效荧光寿命约为41 ns;荧光量子产率测试表明该量子点的绝对荧光量子产率约为71%。
实施例7:CsPbBr1.5I1.5钙钛矿量子点的制备。
称取0.5mmol醋酸铅、0.1mmol碳酸铯,然后加入1.5mL油酸、1.5mL 油胺和8mL十八烯,通氮气加热至120℃并保温30分钟,形成透明溶液A,然后升温至170℃;向溶液A中快速注射87μL溴化氢和99μL碘化氢,保温10秒,用冰浴快速冷却至室温;先离心分离再用1mL环己烷洗涤1次,并将沉淀物分散在30mL环己烷中,得到粒径为11.5nm左右的CsPbBr1.5I1.5钙钛矿量子点。
X射线粉末衍射图(图1g)表明该量子点具有良好的结晶性,其衍射峰位置和相对强度与立方相CsPbBr3的PDF标准卡片(JCPDS NO.75-0412)基本一致,属于立方晶系;透射电镜和粒径分布图(图2d1和2d2)表明该量子点分散性好、形貌均一,粒径约为11.5nm;选区电子衍射图片(图2d3)表明该量子点具有良好的晶化;EDS能谱图(图2d4)指明了该量子点存在Cs、Pb、Br、I元素;该量子点的环己烷溶液(钙钛矿量子点的浓度为1mg/mL)在日光下的照片(图 3a7)呈浅红色,在365nm紫外灯下表现出强橙红光发射(图3b7);吸收光谱 (图4g)表明该量子点在紫外到红光区域具有强吸收,吸收边约为630nm;365 nm激发下的荧光发射光谱(图5g)表明该量子点在631nm具有强发光,半峰宽约为29nm;荧光衰减曲线(图6g)表明该量子点的有效荧光寿命约为66ns;荧光量子产率测试表明该量子点的绝对荧光量子产率约为63%。
实施例8:CsPbBrI2钙钛矿量子点的制备。
称取0.5mmol醋酸铅、0.08mmol碳酸铯,然后加入2mL油酸、2mL油胺和4mL十八烯,通氮气加热至120℃并保温30分钟,形成透明溶液A,然后升温至180℃;向溶液A中快速注射58μL溴化氢和132μL碘化氢,保温5秒,用冰浴快速冷却至室温;先离心分离再用1mL环己烷洗涤1次,并将沉淀物分散在 30mL环己烷中,得到粒径为14.0nm左右的CsPbBrI2钙钛矿量子点。
X射线粉末衍射图(图1h)表明该量子点具有良好的结晶性,其衍射峰位置和相对强度介于立方相CsPbBr3(JCPDS NO.75-0412)和正交相CsPbI3(JCPDS NO.74-1970)之间,属于立方晶系;该量子点的环己烷溶液(钙钛矿量子点的浓度为1mg/mL)在日光下的照片(图3a8)呈红色,在365nm紫外灯下表现出强红光发射(图3b8);吸收光谱(图4h)表明该量子点在紫外到红光区域具有强吸收,吸收边约为660nm;365nm激发下的荧光发射光谱(图5h)表明该量子点在660nm具有强发光,半峰宽约为30nm;荧光衰减曲线(图6h)表明该量子点的有效荧光寿命约为75ns;荧光量子产率测试表明该量子点的绝对荧光量子产率约为59%。
实施例9:CsPbI3钙钛矿量子点的制备。
称取0.5mmol醋酸铅、0.1mmol碳酸铯,然后加入1.5mL油酸、1.5mL 油胺和8mL十八烯,通氮气加热至120℃并保温30分钟,形成透明溶液A,然后升温至150℃;向溶液A中快速注射198μL碘化氢,保温20秒,用冰浴快速冷却至室温;先离心分离再用1mL环己烷洗涤1次,并将沉淀物分散在30mL环己烷中,得到粒径为12.8nm左右的CsPbI3钙钛矿量子点。
X射线粉末衍射图(图1i)表明该量子点具有良好的结晶性,其衍射峰位置和相对强度介于立方相CsPbBr3(JCPDS NO.75-0412)和正交相CsPbI3(JCPDS NO.74-1970)之间,属于立方晶系;透射电镜和粒径分布图(图2e1和2e2)表明该量子点分散性好、形貌均一,粒径约为12.8nm;选区电子衍射图片(图2e3) 表明该量子点具有良好的晶化;EDS能谱图(图2e4)指明了该量子点存在Cs、 Pb、I元素;该量子点的环己烷溶液(钙钛矿量子点的浓度为1mg/mL)在日光下的照片(图3a9)呈深红色,在365nm紫外灯下表现出强深红光发射(图3b9);吸收光谱(图4i)表明该量子点在紫外和整个可见光区域具有强吸收,吸收边约为695nm;365nm激发下的荧光发射光谱(图5i)表明该量子点在700nm具有强发光,半峰宽约为37nm;荧光衰减曲线(图6i)表明该量子点的有效荧光寿命约为81ns;荧光量子产率测试表明该量子点的绝对荧光量子产率约为58%。
实施例10:CsPbBr3钙钛矿纳米线的制备。
称取0.5mmol醋酸铅、0.1mmol碳酸铯,然后加入0.75mL油酸、0.75mL 油胺和10mL十八烯,通氮气加热至120℃并保温30分钟,形成透明溶液A,然后降到室温;向溶液A中快速注射173μL溴化氢,保温30分钟;然后加入5mL 环己烷和5mL丙酮离心洗涤,并将沉淀物分散在30mL环己烷中,得到4×300 nm左右的CsPbBr3钙钛矿纳米线。
透射电镜图(图7a)表明该纳米线分散性好、形貌均一,尺寸约为4×300nm 的纳米线;高分辨透射电镜和选取电子衍射图(图7b和7c)表明该纳米线具有良好的晶化;X射线粉末衍射图(图7d)进一步表明该纳米线具有良好的结晶性,其衍射峰位置和相对强度与立方相CsPbBr3(JCPDS NO.75-0412)基本一致,属于立方晶系;365nm激发下的荧光发射光谱(图7e)表明该纳米线在460nm 和520nm处呈双峰发射,为典型钙钛矿纳米线的特征发射。
实施例11:CsPbCl3:Mn
称取0.5mmol醋酸铅、0.5mmol醋酸锰、0.1mmol碳酸铯,然后加入1mL 油酸、1mL油胺、1mL三正辛基膦和7mL十八烯,通氮气加热至120℃并保温 30分钟,形成透明溶液A,然后升温至180℃;向溶液A中快速注射124μL盐酸,保温10秒;先离心沉淀再用5mL环己烷和5mL丙酮离心洗涤,并将沉淀物分散在30mL环己烷中,得到粒径为12.0nm左右的CsPbCl3:Mn
透射电镜图(图8a)表明该量子点分散性好、形貌均一,粒径约为12.0nm;高分辨透射电镜和选取电子衍射图(图8b和8c)进一步表明该纳米线具有良好的晶化;X射线粉末衍射图(图8d)表明该量子点具有良好的结晶性,其衍射峰位置和相对强度与立方相CsPbCl3(JCPDS NO.75-0411)基本一致,属于立方晶系;365nm激发下的荧光发射光谱(图8e)表明该量子点除了在410nm处有 CsPbCl3量子点的发射,在580nm处还观测到Mn
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
一种ABX型全无机钙钛矿纳米晶的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0