

IPC分类号 : C01B17/20I,C01G11/02I,C01G19/00I,C01G21/21I,B82Y40/00I








专利摘要
本发明公开了一种非层状金属硫化物纳米片的制备方法。所述非层状金属硫化物纳米片的制备方法包括如下步骤:将金属盐、硫源溶解于有机溶剂中,混匀反应,得第一浑浊溶液;加热至澄清透明,通入惰性气体,进行光照反应,然后冷却至室温,得第二浑浊溶液;洗涤、离心分离、干燥得非层状金属硫化物纳米片。本发明提供的制备方法原料种类少、工艺简单、条件温和、具有普适性,且纳米片形貌可控。且由该方法制备得到的非层状金属硫化物纳米片厚度小,为1.2‑9nm,具有良好的各向异性和结晶性,以及良好的应用前景。
权利要求
1.一种非层状金属硫化物纳米片的制备方法,其特征在于,包括如下步骤:
将金属盐、硫源溶解于有机溶剂中,混匀反应,得第一浑浊溶液;加热至澄清透明,通入惰性气体,进行光照反应,然后冷却至室温,得第二浑浊溶液;洗涤、离心分离、干燥得非层状金属硫化物纳米片;
所述硫源包括直链烷基硫醇;
所述光照反应包括在强度为 300 mW·cm
所述光照反应的光源选自高压汞灯。
2.根据权利要求1 所述的制备方法,其特征在于,所述金属盐选自金属的醋酸盐或氯化盐。
3.根据权利要求1 所述的制备方法,其特征在于,所述有机溶剂中金属盐的浓度为10-50 mmol·L
4.根据权利要求1 所述的制备方法,其特征在于,所述硫源选自正辛硫醇、正十二硫醇或正十八硫醇。
5.根据权利要求1 所述的制备方法,其特征在于,所述有机溶剂中硫源的浓度为10-50mmol·L
6.根据权利要求1 所述的制备方法,其特征在于,所述有机溶剂为沸点高于300 ℃的有机溶剂。
7.根据权利要求1 所述的制备方法,其特征在于,所述有机溶剂选自油胺、油酸或十八烯中的至少一种。
8.根据权利要求1 所述的制备方法,其特征在于,所述加热温度为60-140℃。
9.根据权利要求1 所述的制备方法,其特征在于,所述干燥温度为60-80℃;所述干燥时间为12-24h。
10.根据权利要求1 所述的制备方法,其特征在于,洗涤时,洗涤剂为非极性溶剂。
11.根据权利要求10所述的制备方法,其特征在于,所述洗涤剂选自甲苯、正己烷或环己烷。
说明书
技术领域
本发明涉及金属硫化物合成领域。更具体地,涉及一种非层状金属硫化物纳米片及其制备方法。
背景技术
金属硫化物是一类常见的半导体。许多常见的金属硫化物具有较宽的带隙,如PbS、Cu2S、CdS等,因此在可见光区或近紫外区有较好的光吸收,加上合适的带边位置,使得这些金属硫化物具有优良高效的光响应活性。将体相的金属硫化物制成纳米片结构时,由于量子尺寸效应,金属硫化物的电子能带结构发生显著变化,在光催化材料、发光材料、传感器、太阳能电池、光电导原件等诸多方面均有着巨大的应用价值。
对于MoS2、SnS2等部分硫化物,其体相即为层状材料,层与层之间靠相对较弱的范德华力连接,因此可以采用物理剥离的方法制成厚度较小、甚至是单层的二维结构。而对于非层状的金属硫化物,只能通过自下而上的方法得到纳米片。传统非层状的金属硫化物的热合成受限于较高的反应温度,难以对反应的动力学因素进行有效调控,不利于构筑具有各向异性结构的纳米晶。此外,热合成过程涉及较多的反应条件,需要进行大量尝试实验,且不同金属硫化物所需条件不同,难以形成普适、有效的合成方法。
因此,需要提供一种温和、简单且具有普适性的非层状金属硫化物纳米片制备方法。
发明内容
本发明第一个目的在于提供一种非层状金属硫化物纳米片的制备方法。该制备方法原料简单、条件温和、工艺步骤少、具有普适性,且可以实现非层状金属硫化物纳米片的形貌调控。
本发明第二个目的在于提供一种非层状金属硫化物纳米片。
为达到上述目的,本发明采用下述技术方案:
本发明第一个方面,提供一非层状金属硫化物纳米片的制备方法,包括如下步骤:
将金属盐、硫源溶解于有机溶剂中,混匀反应,得第一浑浊溶液;加热至澄清透明;通入惰性气体,进行光照反应,然后冷却至室温,得第二浑浊溶液;洗涤、离心分离、干燥得非层状金属硫化物纳米片。
需要说明的是,金属盐与硫源溶解于有机溶剂中,通过超声、搅拌等方式,使两者充分混匀接触。在这一过程中,硫源作为配体,利用其自身的强配位效应,通过与金属离子络合,形成具有良好层状结构的金属络合物,得到第一浑浊溶液。
通过油浴等方式将第一浑浊溶液加热,并进行搅拌混匀,使金属络合盐在有机溶剂中充分溶解,得到澄清透明溶液,其中的层状结构金属络合物作为制备非层状金属硫化物纳米片的前驱体和软模板。
在惰性气体气氛中,通过油浴等方式对澄清透明溶液继续加热,在光照条件下,层状结构的金属络合盐分解,诱导限域其形成非层状金属硫化物纳米片,其在有机溶剂中的溶解度较小,室温下形成了第二浑浊溶液。经过洗涤、离心分离、干燥后可得到非层状金属硫化物纳米片。
优选地,所述金属盐选自金属的醋酸盐或氯化盐;
在实际应用过程中,可以加入两种或两种以上的金属盐,制备得到含有两种及两种以上金属元素的非层状金属硫化物纳米片。
优选地,所述有机溶剂中金属盐的浓度为10-50mmol·L
进一步地,例如,所述有机溶剂中金属盐的浓度包括但不限于15-45mmol·L
优选地,所述硫源包括直链烷基硫醇;
优选地,所述硫源包括但不限于正辛硫醇、正十二硫醇或正十八硫醇;
选用直链烷基硫醇作为配体,利用其自身的强配位效应,通过与金属离子络合,可以形成具有良好层状结构的金属硫醇盐,以作为制备非层状金属硫化物纳米片的前驱体和软模板。
优选地,所述有机溶剂中硫源的浓度为10-50mmol·L
进一步地,例如,所述有机溶剂中硫源的浓度包括但不限于15-45mmol·L
所述有机溶剂为沸点高于300℃的有机溶剂;
优选地,所述有机溶剂选自油胺、油酸或十八烯中的至少一种。
在制备过程中,层状金属络合物的溶解以及非层状金属硫化物纳米片的形成都是在加热情况下进行的,选用高沸点溶剂以避免其大量发挥,影响制备过程。
优选地,所述加热温度为60-140℃。
优选地,所述光照反应为在强度为300mW·cm
本领域技术人员可以理解的是,反应时间与光源的选择以及光强度有关,本领域技术人员可以在不通过创造性劳动的情况下,通过有限次试验得到,因此只要是在可见光照射下进行的光化学反应制备非层状金属硫化物纳米片,都在本发明的保护范围内,包括但不限于使用高压汞灯提供符合要求的光照。
优选地,所述干燥的温度为60-80℃;所述干燥的时间为12-24h。
优选地,洗涤时,洗涤剂为非极性溶剂;
优选地,所述洗涤剂选自甲苯、正己烷或环己烷。
需要说明的是,洗涤剂是能够溶解反应过程中的高沸点有机溶剂的,进而通过离心分离,得到非层状金属硫化物纳米片。
本发明第二个目的在于提供一种由上述制备方法制备得到的非层状金属硫化物纳米片。
优选地,所述金属硫化物纳米片的厚度为1.2-9nm。
由本发明所提供的方法制备得到的非层状金属硫化物纳米片在光催化材料、发光材料、传感器、太阳能电池、光电导原件等诸多方面均有着巨大的应用价值。
本发明的有益效果如下:
(1)本发明提供的制备方法以层状金属硫醇盐作为前驱体和软模板,不需要添加过多额外的试剂,通过光照下的诱导限域,即可得到非层状金属硫化物纳米片,原料简单、条件温和、制备简便,工艺简单。
(2)本发明提供的制备方法能够普适性地合成多种非层状金属硫化物纳米片,因此可以得到并比较不同金属硫化物纳米片的物理化学性质,并将其应用于不同领域。
(3)本发明提供的制备方法采用的是光化学合成策略,可以在较低的温度下实现产物合成,因此更方便调控产物的形貌。
(4)由本发明提供的制备方法得到的非层状金属硫化物纳米片厚度小,为1.2-9nm,且具有良好的各向异性和结晶性以及良好的应用前景。
附图说明
下面结合附图对本发明的具体实施方式作进一步详细的说明。
图1示出本发明实施例1所获得的PbS纳米片的XRD谱图。
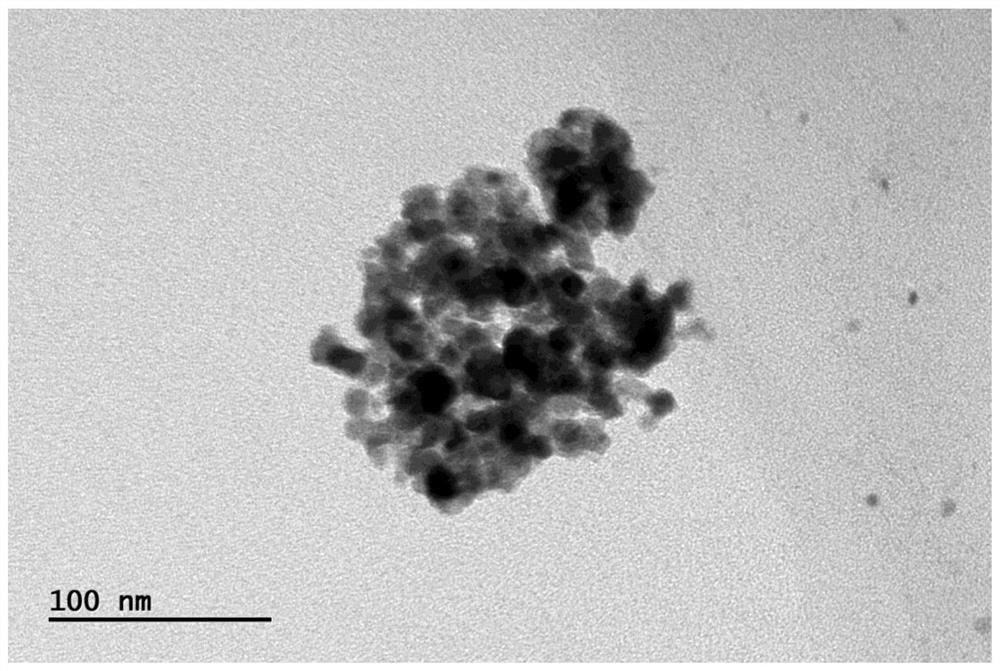
图2示出本发明实施例1所获得的PbS纳米片的透射电镜图。
图3示出本发明实施例1所获得的PbS纳米片的高分辨透射电镜图。
图4示出本发明实施例2所获得的CdS纳米片的XRD谱图。
图5示出本发明实施例2所获得的CdS纳米片的透射电镜图。
图6示出本发明实施例2所获得的CdS纳米片的高分辨透射电镜图。
图7示出本发明实施例3所获得的PbS纳米片的透射电镜图。
图8示出本发明实施例4所获得的PbS纳米片的透射电镜图。
图9示出本发明实施例5所获得的Cu2SnS3纳米片的透射电镜图。
具体实施方式
为了更清楚地说明本发明,下面结合优选实施例和附图对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
本发明中,制备方法如无特殊说明则均为常规方法。所用的原料如无特别说明均可从公开的商业途径获得。
实施例1
一种非层状硫化铅纳米片的制备方法,包括如下步骤:
1)称取0.2mmol醋酸铅、0.4mmol正十二硫醇加入石英烧瓶中,加入5mL油胺和5mL油酸,磁力搅拌至得到浑浊均匀的黄色溶液。
2)将上述溶液在75℃油浴中加热30min,得到澄清透明的黄色溶液。
3)向澄清透明的黄色溶液中通入氩气,在油浴中保持75℃加热的同时,选用500W高压汞灯作为光源,进行4h光照,完毕后自然降温到室温,得到黑色浑浊溶液。
4)在上述黑色浑浊溶液中加入甲苯并混合均匀,以6000转每分钟的转速离心5分钟,该过程重复5次,在60-80℃下干燥12-24h,即得到非层状PbS纳米片。
图1中曲线为实施例1制备的非层状PbS纳米片的XRD谱图。
图2为实施例1制备的非层状PbS纳米片的透射电镜图。
图3为实施例1制备的非层状PbS纳米片的高分辨像透射电镜图。
由图1可知,合成的非层状PbS纳米片XRD谱图中只出现了面心立方相PbS晶体的峰,说明合成的产物是纯相的PbS。与PbS晶体标准峰相比,合成的PbS纳米片仅能观察到两个峰,说明合成的PbS纳米片具有很好的各向异性结构,且厚度很小。由图2可知,合成的PbS纳米片TEM图中只观察片状产物,同样说明合成的PbS为纳米片。由图3可知,合成的PbS纳米片具有良好的结晶性,且通过对晶面间距的测量可以判定该纳米片暴露的晶面为(100)晶面。
实施例2
重复实施例1,其区别仅在于将金属盐由醋酸铅改为氯化镉,油酸改为十八烯,加热温度由75℃改为120℃。
图4中曲线为实施例1制备的CdS纳米片的XRD谱图。
图5为实施例2制备的CdS纳米片的透射电镜图。
图6为实施例2制备的CdS纳米片的高分辨透射电镜图。
由图4可知,合成的CdS纳米片XRD谱图中只出现了CdS晶体的峰,说明合成的产物是纯相的CdS。由图5可知,合成的CdS纳米片TEM图中只观察片状产物,同样说明合成的CdS为纳米片。由图6可知,合成的CdS纳米片具有良好的结晶性,且通过对晶面间距的测量可以判定该纳米片暴露的晶面为(100)晶面。
实施例3-4
重复实施例1,其区别仅在于将硫醇改为辛硫醇或正十八硫醇。得到的PbS为非层状纳米片结构。
图7、8所示为实施例3、4中所合成的PbS纳米片的透射电镜图。图中,采用两种烷基链硫醇所合成的PbS产物均只有非层状纳米片结构,没有出现纳米颗粒或其他形貌产物。
本发明实验证明,在所考察的直链烷基硫醇长度对形成硫化物纳米片没有明显影响。
实施例5
重复实施例1,其区别仅在于将金属盐由醋酸铅改为氯化铜加氯化锡,油胺改为十八烯,加热温度由75℃改为140℃。
得到Cu2SnS3产物为非层状纳米片结构。
图9为所合成的Cu2SnS3纳米片的透射电镜图,图中可以清晰的看到产物是纳米片形貌。
实施例6-7
重复实施例1,其区别仅在于将金属盐由醋酸铅改为氯化铜,氯化铜加氯化铟,油胺改为十八烯,加热温度由75℃改为140℃。
得到的Cu9S5、CuInS2产物均为非层状纳米片结构。
本发明实验证明,该非层状金属硫化物纳米片的合成方法具有广泛的普适性,且能够合成三元的金属硫化物纳米片。
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。
一种非层状金属硫化物纳米片及其制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。

















动态评分
0.0