





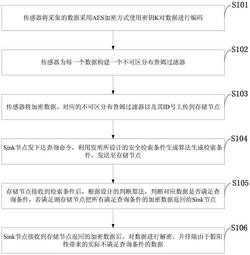




专利摘要
本发明公开了一种定量分析金属嵌入碳纳米管(MCNT)催化剂析氢活性的方法。本发明方法是基于量子化学密度泛函理论模拟来定量分析MCNT催化剂的析氢活性。通过对MCNT模型的稳定性研究,确定模型在析氢反应过程的中稳定存在性;根据吸附位筛选,确定催化剂表面析氢反应发生的具体位置;再结合氢吸附自由能和析氢反应路径计算,定量分析出MCNT催化剂的析氢活性。该方法包括MCNT催化剂模型的构建、模型结构的优化、稳定性计算、吸附位筛选、氢吸附自由能和析氢反应路径计算以及析氢活性分析和表征步骤。本发明无需进行实际实验和催化剂的实际合成即可对MCNT催化剂的析氢活性进行定量分析。
权利要求
1.一种定量分析金属嵌入碳纳米管催化剂析氢活性的方法,其特征在于包括如下步骤:
a催化剂模型构建
根据碳纳米管CNT模型参数,将CNT模型导入到Materials Studio 7.0中,在CNT上制造出缺陷,将金属原子M嵌入到缺陷中,构建出MCNT模型;为充分考虑水分子间氢键的作用,通过加入若干个水分子建立水层模型;将水层模型和MCNT模型进行模型对接,然后将整个模型放到真空层厚度最小为
b模型结构优化
模型结构优化是通过量子化学密度泛函理论计算对初始构建的模型进行结构优化,得到稳定的结构;析氢反应过程包括沃尔默反应、海洛夫斯基反应和塔菲尔反应三个基元反应;模型结构优化涉及析氢反应过程中可能的反应物、中间体和产物,包括原子、自由基和分子;同时计算还涉及到各基元反应初态IS和末态FS中相关物种的共吸附结构;结构优化过程中电子交换函数采用广义梯度近似GGA PBE和DNP基组,收敛标准是0.00001Ha;
c稳定性计算
稳定性计算涉及MCNT催化剂模型的形成能、声子色散谱和第一性原理分子动力学计算;形成能是将凝聚态材料分解成孤立的单原子时平均到每个原子所需要克服的能量;当材料的形成能小于零时,才可以形成稳定的结构,并且形成能越负表示结构越稳定;声子色散谱反映的是声子能量与动量的关系,当声子震动的频率都是实频时表明结构处于相关势能面的局域最小点,对应的是稳定结构;第一性原理分子动力学是将密度泛函理论和分子动力学结合的方法,它可以将分子动力学中的温度和密度泛函理论计算的结构结合起来,通过观测材料原子间键长的变化来预测材料在相应温度下的结构状态,若键长在平衡位置上下波动,则证明结构可以稳定存在,若键长偏离平衡位置,则证明结构稳定性被破坏;
d吸附位筛选
基于结构优化后的吸附质,如水合质子、氢原子和氢分子,在MCNT表面的稳定吸附结构,确定吸附质在MCNT表面的吸附位,并计算各个吸附结构的能量,通过计算吸附结构的能量与吸附前吸附质能量和MCNT能量的差值,得到吸附质在此吸附位的吸附能;吸附能越负的吸附位越容易吸附吸附质,越有利于析氢反应的发生;通过比较各吸附位的吸附能大小,确定最优吸附位,即析氢反应在MCNT催化剂表面发生的最优位置;
e氢吸附自由能和析氢反应路径计算
通过用氢吸附结构的自由能减去吸附前氢原子即吸附质自由能和MCNT自由能计算得到氢吸附自由能;析氢反应路径涉及沃尔默反应、海洛夫斯基反应和塔菲尔反应三个基元反应;从反应初态和末态的稳定结构出发,对各个反应的初态、末态进行原子配对,然后进行反应过渡态TS的搜索计算,求得各个反应中的过渡态,再对初态、过渡态、末态进行能量计算;对初态、过渡态、末态进行频率计算,对求得的能量值进行零点能和熵变修正,最终得到整个反应路径网络及其反应中的结构和能量变化;
f催化剂析氢活性分析与表征
依据氢吸附自由能计算数据,氢吸附自由能越正,越不利于析氢反应过程中的沃尔默反应的发生,氢吸附自由能越负,越不利于析氢反应过程中的海洛夫斯基反应和塔菲尔反应的发生;因此,氢吸附自由能越接近于零,MCNT催化剂析氢活性越高;通过反应路径,确定反应能垒,再结合析氢反应路径网络,确定控制析氢反应速率的速率控制步骤;反应能垒越低,反应越容易进行,对应MCNT催化剂的析氢活性越高。
2.根据权利要求1所述的定量分析金属嵌入碳纳米管催化剂析氢活性的方法,其特征在于:所述的表征手段为形成能、声子色散谱、第一性原理分子动力学、氢吸附自由能以及析氢反应路径。
3.根据权利要求1所述的定量分析金属嵌入碳纳米管催化剂析氢活性的方法,其特征在于:所述的金属M为Ti、V、Cr、Mn、Fe、Co、Ni、Cu、Zn。
4.根据权利要求1所述的定量分析金属嵌入碳纳米管催化剂析氢活性的方法,其特征在于:所述的碳纳米管CNT为CNT(3,3)、CNT(5,5)、CNT(7,7)和CNT(9,9)型碳纳米管。
说明书
技术领域
本发明涉及碳纳米管材料的析氢活性测定,具体涉及这一种定量分析金属嵌入碳纳米管(MCNT)催化剂析氢活性的方法。
背景技术
化石能源是不可再生能源,并且化石能源的燃烧会产生二氧化碳和粉尘,是导致全球变暖和环境污染的主要因素。氢能作为清洁能源,可以通过析氢反应实现氢气的再生。目前制氢的方法主要有煤气化制氢技术、天然气制氢技术、生物质制氢技术和电解水制氢技术。相比于其它制氢技术,电解水制氢技术清洁无污染、产量稳定并且制得的氢气纯度高,因此在商业应用中多采用电解水制氢技术。电解水制氢技术是在水中通入电流将水分子分解成氢气和氧气的过程,为降低电量消耗同时提高氢气产量,需要设计稳定高效的电极催化剂。目前,对电极催化剂的析氢活性进行评价的方法主要是实验方法,即先通过实验方法合成催化剂,然后进行析氢实验测试。但是该方法实验操作环境要求苛刻,实验仪器复杂,实验费用昂贵,并且对新催化剂选择和制备具有盲目性。
随着计算机技术的发展,量子化学计算模拟被越来越多地应用于新材料的设计与研究,成为一种与实验方法并重的研究手段。它可以对催化剂表面发生的反应进行模拟计算,分析催化剂的催化活性。量子化学计算模拟方法在量子力学理论基础上,只需进行计算机模拟计算,无需进行真实实验,效率高、成本低、计算周期短、可重复性高、结果准确,具有普遍指导意义。这使得量子化学计算模拟技术不仅可预测新材料的性能,还具有实验方法所不具有的高效性,可以指导催化剂的设计、制备以及应用。然而,目前国际上还没有利用量子化学计算模拟方法对金属嵌入碳纳米管(MCNT)催化剂的析氢活性进行详细测定和表征的研究案例。
发明内容
本发明目的在于克服实验技术的缺陷和不足,提供一种定量分析金属嵌入碳纳米管(MCNT)催化剂析氢活性的方法。该方法基于量子化学计算中的密度泛函理论计算,来表征和测定MCNT催化剂的析氢活性。通过形成能、声子色散谱、第一性原理分子动力学的计算,确定设计的MCNT催化剂的稳定性;通过吸附能的计算,定量分析析氢反应发生的活性位点;通过析氢反应路径的计算,并结合氢吸附自由能,分析MCNT催化剂的析氢活性。本发明的目的通过以下技术方案实现:
一种定量分析金属嵌入碳纳米管催化剂析氢活性的方法,包括以下步骤:
(1)催化剂模型构建
根据碳纳米管(CNT)模型参数,将CNT模型导入到Materials Studio 7.0中,在CNT上制造出缺陷,将金属原子(M)嵌入到缺陷中,构建出MCNT模型。为充分考虑水分子间氢键的作用,通过加入若干个水分子建立水层模型。将水层模型和MCNT模型进行模型对接,然后将整个模型放到真空层厚度最小为 的周期性盒子中,得到模拟计算的金属嵌入碳纳米管(MCNT)催化剂模型。按照上述方法,依次建立出水合质子、氢原子和氢分子等吸附质在MCNT表面的吸附结构模型。
(2)模型结构优化
模型结构优化是通过量子化学密度泛函理论计算对初始构建的模型进行结构优化,得到稳定的结构。析氢反应过程包括沃尔默反应、海洛夫斯基反应和塔菲尔反应三个基元反应。模型结构优化涉及析氢反应过程中可能的反应物、中间体和产物,包括了原子、自由基和分子;同时计算还涉及到各基元反应初态(IS)和末态(FS)中相关物种的共吸附结构。结构优化过程中电子交换函数采用广义梯度近似(GGA)PBE和DNP基组,收敛标准是0.00001Ha。
(3)稳定性计算
稳定性计算涉及MCNT催化剂模型的形成能、声子色散谱和第一性原理分子动力学计算。形成能是将凝聚态材料分解成孤立的单原子时平均到每个原子所需要克服的能量。当材料的形成能小于零时,才可以形成稳定的结构,并且形成能越负表示结构越稳定。声子色散谱反映的是声子能量与动量的关系,当声子震动的频率都是实频时表明结构处于相关势能面的局域最小点,对应的是稳定结构。第一性原理分子动力学是将密度泛函理论和分子动力学结合的方法,它可以将分子动力学中的温度和密度泛函理论计算的结构结合起来,通过观测材料原子间键长的变化来预测材料在相应温度下的结构状态,若键长在平衡位置上下波动,则证明结构可以稳定存在,若键长偏离平衡位置,则证明结构稳定性被破坏。
(4)吸附位筛选计算
基于结构优化后的吸附质(如水合质子、氢原子和氢分子)在MCNT表面的稳定吸附结构,确定吸附质在MCNT表面的吸附位,并计算各个吸附结构的能量,通过计算吸附结构的能量与吸附前吸附质能量和MCNT能量的差值,得到吸附质在此吸附位的吸附能。吸附能越负的吸附位越容易吸附吸附质,越有利于析氢反应的发生。通过比较各吸附位的吸附能大小,确定最优吸附位,即析氢反应在MCNT催化剂表面发生的最优位置。
(5)氢吸附自由能和析氢反应路径计算
通过用氢吸附结构的自由能减去吸附前氢原子(即吸附质)自由能和MCNT自由能计算得到氢吸附自由能。析氢反应路径涉及沃尔默反应、海洛夫斯基反应和塔菲尔反应三个基元反应。从反应初态和末态的稳定结构出发,对各个反应的初态、末态进行原子配对,然后进行反应过渡态(TS)的搜索计算,求得各个反应中的过渡态,再对初态、过渡态、末态进行能量计算。对初态、过渡态、末态进行频率计算,对求得的能量值进行零点能和熵变修正,最终得到整个反应路径网络及其反应中的结构和能量变化。
(6)催化剂析氢活性分析与表征
依据氢吸附自由能计算数据,氢吸附自由能越正,越不利于析氢反应过程中的沃尔默反应的发生,氢吸附自由能越负,越不利于析氢反应过程中的海洛夫斯基反应和塔菲尔反应的发生。因此,氢吸附自由能越接近于零,MCNT催化剂析氢活性越高。通过反应路径,确定反应能垒,再结合析氢反应路径网络,确定控制析氢反应速率的速率控制步骤。反应能垒越低,反应越容易进行,对应MCNT催化剂的析氢活性越高。
本发明的优点和效果:本发明无需进行实际的实验,克服现有实验技术对析氢实验设备及相应操作复杂以及实验合成新型析氢催化剂盲目性的问题。本发明通过运用基于量子化学密度泛函理论的方法,对析氢反应过程中的反应路径进行模拟计算,并结合反应过程中的氢吸附自由能和反应能垒,对新型纳米管催化剂的析氢活性进行系统分析。
附图说明
图1是本发明定量分析金属嵌入碳纳米催化剂析氢活性的详细步骤图。
图2是模型建立过程,其中(a)是MnCNT(5,5)模型建立过程;(b)是水层模型;(c)是MnCNT(5,5)催化剂模型。
图3是结构优化后的稳定结构,其中(a)是MnCNT(5,5)的俯视图和侧视图;(b)、(c)、(d)、(e)和(f)是单个氢原子可能的吸附结构;(g)、(h)、(i)、(j)和(k)是在第一个氢原子确定吸附位之后,第二个氢原子可能的吸附结构;(l)和(m)分别是MnCNT(5,5)催化析氢过程中沃尔默反应的初态和末态结构;(n)和(o)分别是MnCNT(5,5)催化析氢过程中海洛夫斯基反应的初态和末态结构;(p)和(q)分别是MnCNT(5,5)催化析氢过程中塔菲尔反应的初态和末态结构。
图4是MnCNT(5,5)稳定性计算数据,其中(a)是MnCNT(5,5)的声子色散谱;(b)、(c)和(d)分别是在300K、500K、800K下第一性原理分子动力学模拟的1ps内Mn-C键长的变化趋势。
图5是吸附位筛选计算模型,其中(a)表示单个氢原子所有可能的吸附位,箭头表示在结构优化过程中氢原子发生的转移情况;(b)表示在T吸附位已经吸附一个氢原子后,第二个氢原子所有可能的吸附位,箭头表示在结构过程中氢原子发生的转移情况。
图6是析氢反应路径,其中(a)是析氢反应路径网络;(b)是沃尔默反应路径;(c)海洛夫斯基反应路径;(d)是塔菲尔反应路径。
图7是MnCNT的碳纳米管(CNT)曲率与氢吸附自由能关系图。
图8是嵌入金属种类(M)与MCNT氢吸附自由能关系图。
具体实施方式
本发明定量分析金属嵌入碳纳米管催化剂的析氢活性的方法的详细步骤如图1所示,下面以金属锰(Mn)嵌入(5,5)型碳纳米管(MnCNT(5,5))为例详细说明本发明的催化剂模型构建、模型结构优化、稳定性计算、吸附位筛选、氢吸附自由能和析氢反应路径计算以及催化剂析氢活性分析与表征。应理解,下面内容仅用于说明本发明而不用于限制本发明的范围。
实施例1定量分析金属嵌入碳纳米管MnCNT(5,5)催化剂的析氢活性
(1)MnCNT(5,5)催化剂模型构建
将(5,5)型碳纳米管(CNT(5,5))模型导入到Material Studio 7.0中,在CNT(5,5)模型的基础上,去除碳纳米管切向上两个相邻的碳原子,然后在产生的缺陷中嵌入Mn原子,构建出锰嵌入碳纳米管(MnCNT(5,5))模型,建立过程如图2(a)所示。为充分考虑水分子间的氢键作用,建立如图2(b)所示的水层模型,图中虚线表示水分子间的氢键作用。将水层模型完全覆盖在MnCNT(5,5)模型的金属中心上方,完成水层模型与MnCNT(5,5)模型的对接,在将整个模型放到真空层厚度最小为 的周期性盒子中,得到模拟计算的MnCNT(5,5)催化剂模型,最终模型如图2(c)所示。按照上述方法,依次建立出水合质子、氢原子和氢分子等吸附质在MnCNT(5,5)催化剂表面的吸附结构模型。
(2)模型结构优化
对步骤(1)中所有建立的模型,运用Material Studio 7.0的Dmol
(3)稳定性计算
从结构优化的MnCNT(5,5)(图3(a))出发,对其进行单点能计算,再对自由状态的锰原子和碳原子进行单点能计算,得到各自的能量,求得MnCNT(5,5)的形成能是-7.54eV,即为将MnCNT(5,5)破坏成单个原子所需要的能量,这个数是远大于在其表面上发生析氢反应多需要的能量,说明MnCNT(5,5)在反应条件下是稳定的。
图4(a)是MnCNT(5,5)的声子色散谱,声子振动的频率都是实频(即频率为正,大于零)表明结构处于相关势能面的局域最小点,对应的MnCNT(5,5)是稳定结构。图4(b)、图4(c)和图4(d)分别是在300K、500K、800K下第一性原理分子动力学模拟的1ps内MnCNT(5,5)中Mn-C键长的变化趋势图。可以看出在300K、500K、800K时Mn-C键长均稳定在 上下波动,表明Mn-C键在300K、500K、800K时均没有发生断裂,MnCNT(5,5)结构是稳定存在的。
(4)吸附位筛选
从图3中MnCNT(5,5)表面上各种可能吸附态模型出发,分别进行单点能计算,用求得的能量分别计算出各吸附位对应的吸附能。图5(a)中表示单个氢原子在MnCNT(5,5)表面上所有可能的吸附位。箭头表示在结构优化过程中,氢原子发生的转移情况。计算表明在结构优化过程中MnCNT(5,5)表面上B1、B3、B4吸附位上的氢原子会转移到T2吸附位,H1吸附位的氢原子会转移到T1吸附位,B2吸附位的氢原子会转移到T3吸附位,B5吸附位的氢原子会转移到T4吸附位,最终得到如图3所示的单个氢原子在MnCNT(5,5)表面上所有可能的吸附态,计算求得各吸附位对应的吸附能如表1所示。
表1单个氢原子在MnCNT(5,5)表面上各吸附位的吸附能
吸附能越负,表示氢原子更容易也更可能吸附在该吸附位,从表1中可以看出,MnCNT(5,5)表面上T2吸附位是单个氢原子的最优吸附位。基于单原子最优吸附位(图5(b)中T吸附位)吸附一个氢原子后,图5(b)给出了第二个氢原子在MnCNT(5,5)表面上所有可能的吸附位,箭头表示在结构优化过程中氢原子发生的转移情况。计算表明在结构优化过程中MnCNT(5,5)表面上B2吸附位上的氢原子会转移到T2吸附位,H1和B1吸附位上的氢原子会转移到T1吸附位,B3和H2吸附位的氢原子转移到T3吸附位,最终得到如图3所示所有可能的双氢原子吸附态,计算求得各吸附位对应的吸附能如表2所示。从表2中可以看出,在第一个氢原子吸附在MnCNT(5,5)的T吸附位后,第二个氢原子最可能吸附在T5吸附位上。
表2第二个氢原子在MnCNT(5,5)表面上各吸附位的吸附能
(5)氢吸附自由能和析氢反应路径计算
分别从图3中MnCNT(5,5)催化剂表面上析氢过程的沃尔默反应、海洛夫斯基反应和塔菲尔反应的初态和末态结构出发,计算搜索反应中的过渡态,再对过渡态进行能量计算,最终得到整个析氢反应路径。沃尔默反应是水层中的一个水合质子从水分子中解离下来,吸附到MnCNT(5,5)催化剂表面的过程;海洛夫斯基反应是水层中一个水合质子和MnCNT(5,5)表面上吸附的氢原子结合反应生成氢气的过程;塔菲尔反应是两个水合质子从水分子中解离下来同时吸附在催化剂表面,共同脱附结合生成氢气的过程。图6(a)表示析氢反应的反应路径网络,分为两条反应路径,即沃尔默-海洛夫斯基反应路径和沃尔默-塔菲尔反应路径。(b)为MnCNT(5,5)表面上的沃尔默反应路径,其反应能垒为0.61eV;(c)为MnCNT(5,5)表面上的海洛夫斯基反应路径,其反应能垒为0.80eV;(d)为MnCNT(5,5)表面上的塔菲尔反应路径,其反应能垒为1.72eV。此外,计算表明氢原子在MnCNT(5,5)表面上的吸附自由能为+0.118+eV。
(6)MCNT催化剂析氢活性分析与表征
理论上吸附自由能越接近于零,催化剂的析氢活性越高。MnCNT(5,5)的氢吸附自由能为0.118eV,比较接近于零,说明MnCNT(5,5)具有高析氢活性。另一方面,MnCNT(5,5)表面上沃尔默反应、海洛夫斯基反应和塔菲尔反应的反应能垒分别为0.61eV、0.80eV和1.72eV,表明MnCNT(5,5)催化剂表面有利于沃尔默-海洛夫斯基反应的发生,是析氢反应的主要路径,其速率控制步骤为海洛夫斯基反应,反应能垒仅为0.80eV,进一步说明了MnCNT(5,5)催化剂表面容易发生析氢反应,MnCNT(5,5)催化剂的析氢活性非常高。
表3不同曲率的MnCNT析氢反应能垒(单位:eV)
与MnCNT(5,5)的计算分析过程类似,进一步计算了具有不同CNT曲率的MnCNT催化剂的氢吸附自由能和析氢反应路径,分析了CNT曲率对MnCNT催化剂析氢活性的影响。Mn嵌入的(3,3)、(5,5)、(7,7)和(9,9)型碳纳米管分别记为MnCNT(3,3)、MnCNT(5,5)、MnCNT(7,7)和MnCNT(9,9)。表3给出了MnCNT(3,3)、MnCNT(5,5)、MnCNT(7,7)和MnCNT(9,9)的氢吸附自由能和析氢反应能垒。图7给出来了不同曲率的MnCNT的氢吸附自由能。由图7可知,MnCNT(3,3)、MnCNT(5,5)、MnCNT(7,7)和MnCNT(9,9)的氢吸附自由能分别为+0.363eV、+0.118eV、+0.184eV和+0.190eV,说明随着碳纳米管曲率的增加,其氢吸附自由能呈现“V”型曲线关系,即氢吸附能呈现先减小后增大的趋势,其中MnCNT(5,5)的氢吸附自由能最小,也最接近零。这表明,随着碳纳米管曲率的增加,MnCNT的析氢活性呈现“火山”型曲线关系,即MnCNT析氢活性呈现先增大后减小的趋势,其中MnCNT(5,5)的析氢活性最高。此外,表3给出了不同曲率的MnCNT析氢反应能垒。由表3可以看出,与MnCNT(5,5)表面上的析氢反应路径类似,MnCNT(3,3)、MnCNT(7,7)和MnCNT(9,9)的析氢过程的主要路径也是沃尔默-海洛夫斯基反应,其速率控制步骤为海洛夫斯基反应,对应的反应能垒分别为1.87eV、1.08eV和1.27eV。随着碳纳米管曲率的增加,MnCNT的析氢反应能垒也呈现“V”型曲线关系,析氢反应能垒进一步表明相应的MnCNT析氢活性呈现“火山”型曲线关系,其中MnCNT(5,5)的析氢活性最高。
为分析嵌入金属种类对MCNT析氢活性的影响,分别将Ti、V、Cr、Mn、Fe、Co、Ni、Cu、Zn嵌入到(5,5)型碳纳米管,得到TiCNT(5,5)、VCNT(5,5)、CrCNT(5,5)、MnCNT(5,5)、FeCNT(5,5)、CoCNT(5,5)、NiCNT(5,5)、CuCNT(5,5)和ZnCNT(5,5)。图8给出了不同金属嵌入的碳纳米管以及纯Pt的氢吸附自由能。由图中可知,其氢吸附自由能顺序为:纯Pt(-0.090eV)<MnCNT(5,5)(+0.118eV)<FeCNT(5,5)(-0.146eV)<VCNT(5,5)(-0.273eV)<CrCNT(5,5)(-0.351eV)<CoCNT(5,5)(-0.424eV)<CuCNT(5,5)(-0.484eV)<TiCNT(5,5)(-0.540eV)<NiCNT(5,5)(-0.926eV)<ZnCNT(5,5)(-1.103eV)。结果表明纯Pt的氢吸附自由能最小(-0.090eV),相应的析氢活性最高。在各种金属嵌入的碳纳米管中,MnCNT(5,5)的氢吸附自由能仅次于Pt并接近于零,表明在各种金属嵌入的碳纳米管中,MnCNT(5,5)催化剂的析氢活性最高。
表4给出不同金属原子嵌入的MCNT(5,5)催化剂的析氢反应能垒。由表4可以看出,涉及的所有金属原子嵌入的MCNT(5,5)催化剂都遵循沃尔默-海洛夫斯基反应,相应的速率控制步骤为海洛夫斯基反应。TiCNT(5,5)、VCNT(5,5)、CrCNT(5,5)、MnCNT(5,5)、FeCNT(5,5)、CoCNT(5,5)、NiCNT(5,5)、CuCNT(5,5)和ZnCNT(5,5)的析氢反应能垒分别为1.88eV,1.22eV,1.32eV,0.80eV,1.20eV,1.39eV,2.03eV,1.52eV和2.83eV,说明MnCNT(5,5)催化剂的析氢活性最高,与氢吸附自由能分析的结果相一致。
表4不同金属原子嵌入的MCNT(5,5)析氢反应能垒(单位:eV)
一种定量分析金属嵌入碳纳米管催化剂析氢活性的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0