
吡啶类和膦类钌的络合物,其制备和作为催化剂的用途")
IPC分类号 : C07F15/00,C07B31/00,C07C29/143








专利摘要
本发明描述了一类新型的将2-(氨甲基)吡啶类和膦类作为配体的钌(II)络合物,已证实该络合物在通过氢转移使酮类还原为醇类的反应中具有极高的催化活性。使用上述钌络合物,并以2-丙醇作为氢源,可在短时间内由线状和环状烷基芳基,二烷基和二芳基酮类得到相应的醇类,收率很高。如在氢气流(2-3标准大气压)下操作,酮类至醇类的转换率可达100%。如果使用的膦类具有旋光活性,则可由前手性酮化合物作为起始物制得各种具有旋光活性的醇类,此类醇类在制药工业,农用化学品工业及精细化工业中均是重要的中间体。
权利要求
1.通式(1)的钌(II)络合物
[RuXYLmL’] (I)
式中
X,Y,L,L’分别为:
X,相同或不同,并且为卤素或氢
L配体选自包括下列的组:
a)单齿膦,通式为PR1R2R3,式中R1,R2和R3可相同或不同,并且为脂族或芳香族;
b)双齿膦,通式为PR’2(CH2)xPR”2,式中x=2,3或4,其中R’和R”可相同或不同,并且为脂族或芳香族;
c)具有旋光活性的二膦;
且m=1或2,条件为如配体L选自b)或c)组,则m=1;如配体L选自a)组则m=2,并且在这种情况下,配体可相同或不同;
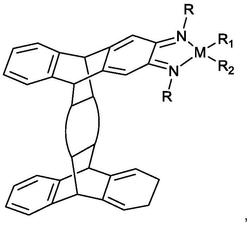
L’为2-(氨甲基)吡啶型双齿配体,分子式为(II)
式中R4,R5可相同或不同,并且可以是H,脂族或芳香族。
2.如权利要求1中所述的钌(II)络合物,其中X和Y配体可为反式或顺式。
3.如权利要求1或2中所述的钌(II)络合物,其中所述的钌(II)络合物的分子式(III)为
反式-[RuXYL2L’] (III)
式中X,Y可单独为卤素或氢,
L为相同或不同,并且为选自a)组的单齿膦类,
L’为分子式为(II)的2-(氨甲基)吡啶型双齿配体,;
4.如权利要求1或2中所述的钌(II)络合物,其中所述的钌(II)络合物的分子式(III)为
顺式-[RuXYL2L’] (III)
式中X,Y可单独为卤素或氢,
L为相同或不同,并且为选自a)组的单齿膦类,
L’为分子式为(II)的2-(氨甲基)吡啶型双齿配体,;
5.如权利要求3或4中所述的钌(II)络合物,其中X和Y可单独为氯或氢,L为单齿膦PPh3和L’为2-(氨甲基)吡啶。
6.如权利要求1或2中所述的钌(II)络合物,其中所述的钌(II)络合物的分子式(IV)为
反式-[RuXYL1L’] (IV)
式中X,Y可单独为卤素或氢,
L为选自b)组的双齿膦或选自c)组的具有旋光活性的二膦,
L’为分子式为(II)的2-(氨甲基)吡啶型双齿配体。
7.如权利要求1或2中所述的钌(II)络合物,其中所述的钌(II)络合物的分子式(IV)为
顺式--[RuXYL1L’] (IV)
式中X,Y可单独为卤素或氢,
L为选自b)组的双齿膦或选自c)组的具有旋光活性的二膦,
L’为分子式为(II)的2-(氨甲基)吡啶型双齿配体。
8.如权利要求6或7中所述的钌(II)络合物,其中X和Y可为氯,L选自包括PPh2(CH2)2PPh2,PPh2(CH2)3PPh2,PPh2(CH2)4PPh2的组中的二膦,且L’为2-(氨甲基)吡啶。
9.如权利要求6或7中所述的钌(II)络合物,其中X和Y可为氯,L选自包括下列的组中的具有旋光活性的二膦:
(2S,3S)-(-)-2,3-双-(二苯膦基)丁烷(CHIRAPHOS),(S)-(-)-2,2’-双-(二-p-甲基苯膦基)-1,1’-联萘(Tol-BINAP),(2S,4S)-(-)-2,4-双-(二苯膦基)戊烷(SKEWPHOS),(4R,5R)-(-)-O-异亚丙基-2,3-二羟基配位-1,4-双(二苯膦基)丁烷(DIOP),(R)-(-)-1-[(S)-2-(二苯膦基)三茂铁基]乙基二环己基膦(JOSIPHOS),L’为2-(氨甲基)吡啶。
10.如上述任一权利要求所述的钌(II)络合物的合成方法。
11.如上述任一权利要求所述的钌(II)络合物的原位合成方法。
12.通式(I)的钌(II)络合物
[RuXYLmL’] (I)
式中
X,Y,L,L’分别为:
X,Y可相同或不同,并且为卤素或氢
L配体选自包括下列的组:
a)单齿膦,通式为PR1R2R3,式中R1,R2和R3可相同或不同,并且为脂族或芳香族;
b)双齿膦,通式为PR’2(CH2)xPR”2,式中x=2,3或4;R’和R”可相同或不同,并且为脂族或芳香族;
c)具有旋光活性的二膦;
且m=1或2,条件为如配体L选自b)或c)组,则m=1;如配体L选自a)组则m=2,并且在这种情况下,配体可相同或不同;
L’为分子式为(II)的2-(氨甲基)吡啶型双齿配体,
式中R4,R5可相同或不同,并且可以是H,脂族或芳香族,所述的钌(II)络合物原位生成。
13.如上述任一权利要求所述的钌(II)络合物用作催化剂的用途,通过将醇上的氢转移到所述酮类上使酮类进行光学异构选择性和非光学异构选择性还原以制备与所述酮类相应的醇类。
14.如上述任一权利要求所述的钌(II)络合物用作催化剂的用途,通过与氢气反应使酮类进行光学异构选择性和非光学异构选择性还原以制备与所述酮类相应的醇类。
15.如上述任一权利要求所述的钌(II)络合物在反应中用作催化剂的用途,所述反应中通过将醇上的氢转移到所述酮类的还原反应和用气态氢的氢化还原反应两者相结合,使酮类进行光学异构选择性或非光学异构选择性还原以获得相应的醇类。
说明书
技术领域技术领域
本发明述及一类具有2-(氨甲基)吡啶类和膦类的钌(II)络合物,在某些情况下可以是手性的,以及它们通过氢转移法将酮类还原为醇类作为催化剂的用途。在低压(2-3大气压)氢气流下操作,还原率和醇收率可得到进一步的提高。
技术背景背景技术
将羰基化合物还原为醇类是一类具有广泛应用价值的反应,并在近年来研发出一系列催化方法,试图替代传统的化学计量还原体系。在氢气流下使用基于过渡金属(铱,铑,钯,镍)的催化体系进行还原取得了不错的效果,不过最近研究的焦点集中在更具活性的钌衍生物上了。在碱性环境中[RuCl2(膦)2(1,2-双胺)]和[RuCl2(二膦)(1,2-双胺)]型化合物对于匀相中的各类酮的选择性氢化都是非常好的催化剂。此外,使用适当结合的手性双膦和双胺,可对羰基化合物进行光学异构氢化作用以生成具有旋光活性的醇,其对映体过量值(enantiomeric excesses)接近100%。这些反应一般在加压氢和中等温度条件下进行(R.Noyori,Asymmetric Catalysis in Organic Synthesis,Ed.R.Noyori,1994,pp.56-82;T.Ohkuma,H.Ooka,T.Ikariya,R.Noyori,J.Am.Chem.Soc.1995,117,10417;R.Noyori,T.Ohkuma,Angew.Chem.,Int.Ed.Engl.2001,40,40;K.V.L.Crépy,T.Imamoto,Adv.Synth.Catal.2003,345,79)。在氢气流下进行还原反应具有一定的危险性,另一类可供选择并经证实的方法是基于氢转移反应法的催化还原方法。在这类方法中,一般使用2-丙醇作为氢源和反应溶剂,其优点是沸点低,毒性小,环境污染少(G.Zassinovich,G.Mestroni,S.Gladiali,Chem.Rev.1992,92,1051;R.Noyori,S.Hashiguchi,Acc.Cbem.Res.1997,30,97;J.-E.Bckvall,J.Organomet.Chem.2002,652,105;M.J.Palmer,M.Wills,Tetrahedron:Asymmetry 1999,10,2045)。由于氢转移催化法操作简便,并且结果好,因此主要用在小至中等规模的反应中,以取代氢分子还原法。曾使用基于铑和铱等过渡金属的催化剂(M.J.Palmer,M.Wills,1999 ref.cit.;A.J.Blazer,B.J.Mellor,United States Patent 6,372,931,2002 and 6,545,188,2003;A.C.Hillier,H.M.Lee,E.D.Stevens,S.P,Nolan,Organometallics 2001,20,4246)但钌衍生物尤其对于酮类的光学异构选择性还原取的结果最佳。这些衍生物包括:具有二膦-双胺和二膦-二亚胺型作为四配位基的配体的络合物((J.-X.Gao,T.Ikarya,R.Noyori,Organometallics,1996,15,1087),具有二胺基或β-氨基醇为配体的芳烃-钌络合物((K.-J.Haack,S.Hashiguchi,A.Fujii,T.Ikariya,R.Noyori,Angew.Chem.Int.Ed.Engl.1997,36,285;T.Ikariya S.Hashiguchi,J.Takehara,N.Uematsu,K.Matsumara,R.Noyori,A.Fujii,United States Patent,6,184,381,2001;K.Everaere,A.Mortreaux,J.Carpentier,Adv.Synth.Catal.2003,345,67),具有唑啉作为配体的络合物((Y.Jiang,Q.Jiang,X,Zhang,J.Am.Chem.Soc.1998,120,3817;X.Zhang,United States Patent,6,451,727,2002),具有唑啉基三茂铁基配体的络合物(Y.Nishibayashi,I.Takei,S.Uemura,M.Hidai,Organometallics1999,18,2291)。这样通过使用手性配体,可容易地制得具有旋光活性的醇类,它们广泛地在制药工业,农用化学品工业及精细化工工业上具有重要的用途。这些反应一般在强碱,如碱金属的氢氧化物或醇盐下发生反应,其中底物/催化剂的比率为20-2000,起始酮转化为醇的转化率%非常高,并且光学异构选择性达99%。应注意的是这些催化体系,一般催化活性不高,并且TOF值(转换频率=在转化率为50%时每摩尔催化剂每小时转化为醇的酮的摩尔数/,)一般为102-103h-1。这导致反应时间变长,以及设备利用率降低,并且反应时间过长还可能引起催化剂失活和分解的危险,这些都会大大增加产品成本。
值得注意的是对于酮类的非光学异构选择性还原活性很强的催化剂:一种为vanKoten及其合作者(P.Dani,T.Karlen,R.A.Gossage,S.Gladiali,G.van Koten,Angew.Chem.,Int.Ed.Engl.2000,39,743)研制的螯合芳基型催化剂,分子式为RuX[C6H3(CH2PPh2)2-2,6](PPh3)(X=C1,CF3SO3),并具有一稳定的Ru-C芳基键;另一种为Mathieu发表具有三配位基吡啶配体的催化剂,该催化剂对乙酰苯还原反应的TOF值达90000h-1((H.Yang,M.Alvarez,N.Lugan,R.Mathieu,J.Chem.Soc.,Chem.Commun.1995,1721;H.Yang,M.Alvarez-Gressier,N.Lugan,R.Mathieu,Organometallics 1997,16,1401),不过这2种催化剂的迅速失活的特点限制了其在有机合成中的应用。此外,在最近的一项研究中,Mathey和Le Floch(C.Thoumazet,M.Melaimi,L.Ricard,F.Mathey,P.Le Floch,Organometallics 2003,22,2580)描述了一种新型的具有N,P二配位基1-(2-甲基吡啶)-2,5二苯膦作为配体的芳烃钌催化剂,该催化剂对于多种酮类的TOF值达106h-1,但是其过长的反应时间(90℃下要数天时间)限制了它的实际应用。
还有报道说通式为RuXY(PR1R2R3)n(NR6R7R8)m[X和Y=H或卤原子;R1-R3=碳氢可能取代的如苯基等;R6-R8=H或可取代的碳氢化合物原子;n和m=0-4]的钌络合物可在氢转移或氢化反应中起催化作用(T.Ikariya,H.Ikehira,K.Murata,N.Kiyofuji,H.Oooka,S.Hashiguchi,T.Okuma,R.Noyori Japanese Patent,11189600)。值得注意的还有,使用在氮上发生了取代的手性2-(氨甲基)吡啶单体和RuCl2(PPh3)3,前体,原位生成的催化体系可用于光学异构选择性氢转移反应。以上这些体系的活性均较低,光学异构选择性为中等(E.Mizushima,H.Ohi,M.Yamaguchi,T.Yamagishi,J.Mol.Catal.A 1999,149,43)或良好(H.Brunner,M.Niemetz,Monatshefte fürChemie 2002,133,115;H.Brunner,F.Henning,M.Weber,Tetrahedron:Asymmetry2002,13,37)。
为使酮醇还原的氢转移法在经济上更有竞争性,首要目标是要研制出比上述几种体系更有活性和生产率更高的催化剂。如果研制出的催化体系能引起光学异构选择性还原反应是非常重要的,从而使它们用于将前手性酮类合成具有旋光活性的醇类。
因此,本发明的一个目的是获得可在不对称和非不对称性酮类还原反应氢转移法中作为高活性催化剂的钌络合物。本发明的又一目的是在氢转移法的不对称和非不对称性酮类还原反应中原位制备用作催化剂的钌(II)络合物。
发明内容发明内容
为达到上述目的,本发明者已经发现了一类新型具有2-(氨甲基)吡啶作为配体的钌(II)络合物,获得具有高催化活性的催化剂的技术方案,并且通过与手性膦适当组合还获得了光学异构选择性催化剂的可能性。该催化剂还可在合成过程中原位制备。需强调的是这里所描述的体系可在低压和室温条件下作为酮类与分子氢的氢化作用中的催化剂。
为此本发明提供了通式(I)的钌(II)络合物
[RuXYLmL’] (I)
式中
X,Y,L,L’分别为:
X,Y可相同或不同,并且为卤素或氢
L配体选自包括下列物质的组:
a)单齿膦,通式为PR1R2R3,式中R1,R2和R3可相同或不同,并且为脂族或芳香族;
b)双齿膦,通式为PR’2(CH2)xPR’’2,式中x=2,3或4;R’和R”可相同或不同,并且为脂族或芳香族;
C)具有旋光活性的二膦;
且m=1或2,条件为如配体L选自b)或c)组,则m=1;如配体L选自a)组则m=2,并且在这种情况下,配体L可相同或不同;
L’为2-(氨甲基)吡啶型双齿配体,分子式为(II)
式中R4,R5可相同或不同,并且可以是H,脂族或芳香族。
本发明的进一步的方面还包括该钌(II)络合物的原位合成方法,以及在酮的还原过程中通过将氢由醇转移至所述酮类用所述的方法原位直接制备的钌(II)络合物。
本发明的进一步方面是所述钌(II)络合物作为氢转移法酮还原反应中的催化剂的用途。
本发明的进一步方面是所述钌(II)络合物作为与气态氢反应还原酮中的催化剂。
可将从醇上转移氢的还原与气态氢的还原结合使用,从而导致酮醇的完全转化。
附图说明具体实施方式通过下面的详细说明,以及几个优选实例,不限于这些实例,可以更清楚地说明本发明上述和其他方面,以及本发明的特点和优点。
详细说明
如前所述,通式为RuXY(PR1R2R3)n(NR6R7R8)m的钌络合物,式中X和Y为卤素或氢,(PR1R2R3)n为单齿膦型(n=2)或双齿膦型(n=1)配体,及(NR6R7R8)m为双胺,可作为氢转移或氢化反应中的催化剂((T.Ikariya et al ref.cit.)。此外,由与母体RuCl2(PPh3)3,结合的2-(RHN-CHR)C5H4N型的手性吡啶配体原位生成的的催化体系已发现在光学异构选择性氢转移反应中具有活性。但是,应强调的是,在第一种情况下,所提到的双齿配体中未包括2-(氨甲基)吡啶类,而2-(氨甲基)吡啶类构成的配体正是本发明的主要目的之一;而在第一种情况下2-(RHN-CHR)C5H4N型的手性吡啶类配体制备的体系的活性较低,其光学异构选择性为中等(E.Mizushima,et al ref.cit.)或良好(H.Brunneret al.ref.cit.)。因此,上述研究并未显示2-(氨甲基)吡啶型的双齿配体与单齿或双齿膦合用,可获得对氢转移反应有极高催化活性的钌络合物。还应强调的是二氯衍生物(X=Y=Cl),其中氯原子为顺式取向,具有较高的催化活性,而这在先前未见记载。
本发明提供的新型钌(II)络合物,可作为氢转移法将酮还原为醇的反应中的催化剂,其中醇可为手性,其通式(I)为
[RuXYLmL’] (I)
式中
X,Y,L,L’分别为:
X,Y可相同或不同,并且为卤素或氢
L配体选自包括下列的组:
a)单齿膦,通式为PR1R2R3,式中R1,R2和R3可相同或不同,并且为脂族或芳香族;
b)双齿膦,通式为PR’2(CH2)xPR’型2,式中x=2,3或4;R’和R”可相同或不同,并且为脂族或芳香族;
c)具有旋光活性的二膦;
且如选自a)组的相同或不同的单齿膦,则m=2,如所述膦选自b)或c)组,则m=1;,L’为2-(氨甲基)吡啶型双齿配体,分子式为(II)
式中R4,R5可相同或不同,并且可以是H,脂族或芳香族。
络合物中的X和Y配体可呈反式或顺式取向。下图给出2例可能的顺式和反式异构体的结构的实例,其中R4=R5=H:
反式 顺式
为达到本发明的目的,可根据X,Y,L,L’和m不同含义的组合,获得钌(II)络合物,其通式分别如下:
-分子式为(III)的反式或顺式钌(II)络合物
反式或顺式-[RuXYL2L’] (III)
式中X,Y可单独为卤素或氢,
L为相同或不同,并且为选自a)组的单齿膦类,
L’为2-(氨甲基)吡啶型双齿配体,分子式为(II);
反式或顺式钌(II)络合物的分子式(IV)
反式或顺式-[RuXYL1L’] (IV)
式中X,Y可单独为卤素或氢,
L为选自b)组的双齿膦或选自c)组的具有旋光活性的二膦,
L’为2-(氨甲基)吡啶型双齿配体,分子式为(II).
为达到本发明的目的,优先选择的X和Y配体包括:氯和氢;a)组中优先选择的L配体为PPh3;b)组中优先选择的L配体为PPh2(CH2)2PPh2,PPh2(CH2)3PPh2,PPh2(CH2)4PPh2;c)组中优先选择的L配体为:(2S,3S)-(-)-2,3-双-(二苯膦基)丁烷(CHIRAPHOS),(S)-(-)-2,2’-双-(二-p-甲基苯膦基)-1,1’-联萘(Tol-BINAP),(2S,4S)-(-)-2,4-双-(二苯膦基)戊烷(SKEWPHOS),(4R,5R)-(-)-O-异亚丙基-2,3-二羟基配位-1,4-双(二苯膦基)丁烷(DIOP),(R)-(-)-1-[(S)-2-(二苯膦基)三茂铁基]乙基二环己基膦(JOSIPHOS);分子式(II)中L’配体的R4和R5的优先选择为H,因此L’配体的优先选择为2-(氨甲基)吡啶。
已经被分离并用于催化剂的钌络合物具体实例如下,但本发明并不限于这些实例:
1.通式(III)的钌络合物
反式-[RuXYL2L’](III)
式中L’为2-(氨甲基)吡啶,L为PPh3,X=Y=Cl(3)或X=H and Y=Cl(4)
2.通式(III)的钌络合物
顺式-[RuXYL2L’](III)
式中L’为2-(氨甲基)吡啶,L为PPh3,且X=Y=Cl(5)或X=Y=H(6)。
3.通式(IV)的钌络合物
反式-[RuXYLL’](IV)
式中L’为2-(氨甲基)吡啶,L为PPh2(CH2)4PPh2,且X=Y=Cl(7)。
4.通式(IV)的钌络合物
顺式-[RuXYLL’](IV)
式中L’为2-(氨甲基)吡啶,X=Y=Cl,L为下列二膦类PPh2(CH2)2PPh2(8),PPh2(CH2)3PPh2(9),和PPh2(CH2)4PPh2中的一种(10)
或者式中L为一种手性二膦,如(2S,3S)-(-)-2,3-双-(二苯基膦基)丁烷(CHIRAPHOS)(11),(2S,4S)-(-)-2,4-双-(二苯基膦基)戊烷(SKEWPHOS)(12),(4R,5R)-(-)-O-异亚丙基-2,3-二羟基-1,4-双(二苯膦基)丁烷(DIOP)(13),(R)-(-)-1-[(S)-2-(二苯膦基)三茂铁基]乙基二环己基膦(JOSIPHOS)(14),(S)-(-)-2,2’-双-(二-p-甲苯膦基)-1,1’-联萘(Tol-BINAP)(15)。
A.钌络合物的合成
本发明中络合物(3-15)的合成使用化合物RuCl2(PPh3)3(1)作起始物,
该化合物可以购买,也可通过水合RuCl3与三苯基膦反应制得(R.Holm,Inorg.Synth.1970,12,238),络合物RuCl2[PPh2(CH2)4PPh2](PPh3)(2)可按文献记载的方法制备(C.W.Jung,P.E.Garrou,P.R.Hoffman,K.G.Caulton,Inorg.Chem.1984,23,726)。
在常温条件下将RuCl2(PPh3)3(1)在二氯甲烷中按1∶1比率与2-(氨甲基)吡啶反应,可制得反式络合物(3);在回流加热条件下,将RuCl2(PPh3)3(1)在甲苯中与2-(氨甲基)吡啶反应可制得顺式络合物(5)。按与络合物(3)相同的制备方法,以络合物(2)为起始物,与2-(氨甲基)吡啶反应,可制得反式衍生物(7)。以络合物(5)为起始物,以1∶1的比率与一合适的二膦反应,可制得反式络合物(8-13)。将络合物(1)与一合适的手性二膦反应,然后加入2-(氨甲基)吡啶可制得络合物(14)和(15)。对于络合物(10),给出另外2种快速合成途径:以络合物(2)为起始物与双胺反应(方法a)及以络合物(1)为起始物与双胺及相应的二膦反应(方法b)。以2-(氨甲基)吡啶和由络合物(1)合成的RuHCl(PPh3)3s为起始物(R.A.Schunn,E.R.Wonchoba,Inorg.Synth.1971,13,131)反应可制得一氢化物络合物(4),而以络合物(4)为起始物,与异丙醇反应可制得二氢化物络合物(6)。
以下详细地说明了络合物(3)-(6),(10),(12)和(14)的合成方法及特性,但本发明不限于这些实例。全部合成方法在氩气中进行,使用蒸馏或先经脱气的溶剂。
实例1:反式-RuCl2(PPh3)2[2-(H2NCH2)C5H4N](3)的合成
将络合物RuCl2(PPh3)3(1)(0.400g,0.417mmol)以5ml蒸馏过的二氯甲烷制成悬浮液,与2-(氨甲基)吡啶(45L,0.436mmol)反应。在常温下将反应混合物搅拌2小时后,溶液体积减半,然后加入5ml戊烷析出络合物。滤出制得的固体物,以10ml乙醚洗涤2次,并减压干燥。
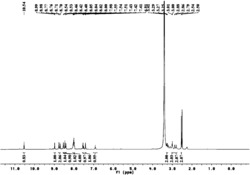
产量250mg(75%)。C42H38Cl2N2P2Ru.的元素分析,计算为(%):C,62.69;H,4.76;N,3.48;发现C,62.85;H,4.80;N,3.54.1H核磁共振(NMR)分析(200.1MHz,CDCl3,20℃,TMS):δ8.53(d,J(HH)=4.2Hz,1H;o-C5H4N),7.60-6.50(m,33H;芳香质子),4.46(宽s,2H;CH2),3.29(宽s,2H;NH2).13C{1H}NMR分析(50.3MHz,CDCl3,20℃,TMS):δ162.8(s;NCCH2),157.6(s;NCH of C5H4N),136.6-120.1(m;芳香C),50.8(s;CH2).31P{1H}NMR分析(81.0MHz,CDCl3,20℃,H3PO4):δ44.0(d,J(PP)=32.7Hz),40.1(d,J(PP)=32.7Hz)。
实例2:络合物反式-RuHCl(PPh3)2[2-(H2NCH2)C5H4N](4)的合成
将络合物RuHCl(PPh3)3(211mg,0.228mmol)以10ml正庚烷制成悬浮液,与2-(氨甲基)吡啶(24L,0.233mmol)反应,然后回流加热1小时。滤出黄色产物,用正庚烷(3×5ml)洗涤,并减压干燥。
产量:118mg(67%).C42H39ClN2P2Ru的元素分析(%),计算为:C,65.49;H,5.10;N,3.64;发现:C,65.23;H,5.03;N,3.41.1H NMR分析(200.1MHz,CD2Cl2,20℃,TMS):δ8.20(s,1H;o-C5H4N),7.70-6.40(m,33H;芳香质子),4.30(假t,J(HH)=14.1Hz,1H;CH2),4.07(d,J(HH)=14.3Hz,1H;CH2),2.87(假t,J(HH)=10Hz,1H;NH2),2.20(假d,J(HH)=10Hz,1H;NH2),-17.70(dd,J(HP)=23.5,29.7Hz).13C{1H}NMR分析(50.3MHz,CD2Cl2,20℃,TMS):δ159.7(s;NCCH2),155.6(d,J(CP)=4.0Hz;NCH),138.8-118.7(m;芳香C),53.4(s;CH2).31P{1H}NMR分析(81.0MHz,CD2Cl2,20℃,H3PO4):δ73.7(d,J(PP)=37.0Hz),68.9(d,J(PP)=37.0Hz)。
实例3:络合物顺式-RuCl2(PPh3)2[2-(H2NCH2)C5H4N](5)的合成
将络合物RuCl2(PPh3)3(1)(1.34g,1.40mmol)以10ml甲苯制成悬浮液,与2-(氨甲基)吡啶(0.160ml,1.55mmol)反应。将反应混合物回流加热2小时,溶液体积减半,再加入5ml戊烷后析出络合物。滤出的固体产物,用5ml乙醚洗涤2次,并减压干燥。
产量:750mg(66.4%).C42H38Cl2N2P2Ru的元素分析(%),计算为:C,62.69;H,4.76;N,3.48;发现:C,62.31;H,4.87;N,3.60.1H NMR分析(200.1MHz,CD2Cl2,20℃,TMS):δ9.16(d,J(HH)=5.7Hz,1H;正-C5H4N),7.70-6.89(m,33H;芳香质子),3.65(m,2H;CHHNHH),3.00(m,1H,CH2),1.42(m,1H,NH2).31P{1H}NMR分析(81.0MHz,CD2Cl2,20℃,H3PO4):δ50.5(d,J(PP)=33.4Hz),43.8(d,J(PP)=33.4Hz)。
实例4:络合物顺式-RuH2(PPh3)2[2-(H2NCH2)C5H4N](6)的合成
将1.4ml异丙醇钠/2-丙醇(0.2 M;0.280mmol)溶液置入Schlenk瓶中,减压蒸去溶剂。加入络合物(4)(211mg,0.274mmol)和甲苯(12ml),制成悬浮液,悬浮液在30℃保温3小时后过滤。减压蒸去甲苯后,制得深红色固体,将该固体减压干燥。
产量:131mg(65%).C42H40N2P2Ru的元素分析(%),计算为:C,68.56;H,5.48;N,3.81;发现:C,68.30;H,5.33;N,3.62.1H NMR分析(200.1MHz,C6D6,20℃,TMS):δ7.93-5.73(m,34H;芳香质子),2.76(t,J(HH)=6.2Hz,2H;CH2),1.67(t,J(HH)=6.0Hz,2H;NH2),-16.31(td,J(HP)=27.5Hz,J(HH)=6.7Hz,1H;RuH),-18.24(td,J(HP)=27.7Hz,J(HH)=6.7Hz,1H;RuH).13C{1H}NMR分析(50.3MHz,C6D6,20℃,TMS):δ158.7(s;NCCH2),155.8(s;NCH),142.0-118.0(m;芳香C),51.4(s;CH2).31P{1H}NMR分析(81.0MHz,C6D6,20℃,H3PO4):δ67.2。
实例5:络合物顺式-RuCl2[PPh2(CH2)4PPh2][2-(H2NCH2)C5H4N](10)的合成(方法a)
将络合物RuCl2[PPh2(CH2)4PPh2](PPh3)(2)(202mg,0.235mmol),以5ml甲苯制成悬浮液,然后与2-(氨甲基)吡啶(27 L,0.262mmol)反应,反应混合物回流加热20小时。加入戊烷后析出沉淀物,过滤后用3ml乙醚洗涤2次,并减压干燥。
产量:126mg(76%).C34H36Cl2N2P2Ru的元素分析(%),计算为:C,57.79;H,5.14;N,3.96;发现:C,57.48;H,5.27;N,3.70.1H-NMR分析(200.1MHz,CDCl3,20℃,TMS):δ9.36(m,1H;正-C4H5N),8.23-6.62(m,23H;芳香质子),4.13(m,1H;CHHP),3.74(m,2H;CHHN,NHH),3.22(m,1H,CHHN),2.82(m,1H,CHHP),2.34-0.90(m,7H;P(CH2)4P,NHH).13C{1H}NMR分析(50.3MHz,CDCl3,20℃,TMS):δ158.0(s;NCCH2),151.1(s;NCH),136.5-119.8(m;芳香C),53.5(s;CH2N),34.8(d,J(CP)=27.0 Hz;CH2P),29.7(d,J(C,P)=29.9Hz;CH2P),27.6(s;CH2),19.7(s;CH2).31P{1H}NMR分析(81.0MHz,CDCl3,20℃,H3PO4):δ54.9(d;J(PP)=37.0Hz),40.1(d;J(PP)=37.0Hz)。
实例6:络合物顺式-RuCl2[PPh2(CH2)4PPh2][2-(H2NCH2)C5H4N](10)的合成(方法b)
将络合物RuCl2(PPh3)3(1)(1.95g,2.03mmol)以30ml甲苯制成悬浮液,与2-(氨甲基)吡啶(0.250mL,2.43mmol)反应,反应混合物在110℃下回流1小时。常温下加入膦PPh2(CH2)4PPh2(853mg,2.00mmol),反应混合物回流20小时。加入戊烷后析出沉淀物,滤出后用3ml乙醚洗涤2次,并减压干燥。
产量:1.25g(87%)
实例7:络合物顺式-RuCl2[(2S,4S)-(-)-2,4-双-(二苯膦基)戊烷][2-(H2NCH2)C5H4N](12)的合成
将络合物顺式-RuCl2(PPh3)2[2-(H2NCH2)C5H4N](5)(303mg,0.377mmol)以5ml甲苯制成悬浮液。混合物回流20小时后,溶液体积减半,加入2ml戊烷后析出络合物。滤出固体沉淀物,并减压干燥。
产量:200mg(74%).C35H38Cl2N2P2Ru的元素分析(%),计算为:C,58.34;H,5.32;N,3.89;发现:C,58.06;H,5.17;N,3.63.1H NMR分析(200.1MHz,CDCl3,20℃,TMS):δ8.78(d,J(HH)=3.1Hz,1H;正-C5H4N),7.95-6.69(m,23H;芳香质子),4.20(宽s,1H;NH),3.61(d,J(HH)=15.6Hz,1H;CHN),3.37(m,1H;PCH),3.07(m,1H;PCH),2.81(宽s,1H;CHN),2.33-1.63(m,2H;CH2),1.25(宽s,1H;NH),1.16(dd,J(HP),J(HH)=7.2,13.6Hz,3H;CH3),0.76(dd,J(HP),J(HH)=7.0,11.6,3H;CH3).13C{1H}NMR分析(50.3MHz,CDCl3,20℃,TMS):δ158.4(s,NCCH2),149.6(s,NCH),139.8-119.3(m,芳香C),51.5(s,CH2N),37.8(s;CH2),33.5(d,J(CP)=27.2Hz;CHP),20.3(d,J(CP)=32.1Hz;CHP),18.9(d,J(CP)=6.6Hz;CH3),17.7(d,J(CP)=1.6Hz;CH3).31P{1H}NMR分析(81.0MHz,CDCl3,20℃,H3PO4):δ64.8(d,J(PP)=44.7Hz),45.3(d,J(PP)=44.7Hz)。
实例8:络合物顺式-RuCl2{(R)-(-)-1-[(S)-2-(二苯膦基)三茂铁基]乙基二环己基膦}[2-(H2NCH2)C5H4N](14)的合成
将络合物RuCl2(PPh3)3(1)(228mg,0.238mmol)和膦(R)-(S)-(-)-Josiphos(141mg,0.237mmol)以10ml甲苯制成悬浮液,回流30分钟,待反应混合物恢复至常温后,加入2-(氨甲基)吡啶(26 L,0.252 mmol)。将该混合物在110℃下再回流4小时。然后用戊烷沉淀,滤出后用3ml乙醚洗涤2次,并减压干燥。
产量:182mg(88%).C42H52Cl2N2P2RuFe的元素分析(%),计算为:C,57.68;H,5.99;N,3.20;发现:C,57.47;H,5.80;N,3.25.1H NMR分析(200.1MHz,CDCl3,20℃,TMS):δ10.25(s,1H;o-H-吡啶),8.39-7.15(m,13H;芳香质子),5.10(m,1H;CHCH3),4.47(m,1H;C5H3),4.30(m,1H;C5H3),3.84-3.34(m,5H;C5H3,CH2NH2)3.66(s,5H;C5H5),2.20-0.52(m,25H;CH3,Cy).31P{1H}NMR分析(81.0MHz,CDCl3,20℃,H3PO4):δ60.8(d,J(PP)=40.9Hz),39.7(d,J(PP)=40.9Hz)。
B.催化试验
本发明的钌(II)络合物可用于氢转移反应法,从相应的酮类制成醇类。在这种新型钌基催化剂及一种碱金属氢氧化物存在下,可将下述酮还原为醇类,其中所述酮为环状酮类,线状二烷基酮类,烷基芳基酮类和二芳基酮类R6C(=O)R7,式中R6和R7代表一饱和或不饱和脂族,或一芳香羟族;可具有或不具有烷取代基,该取代基具有氧,卤原子或一杂环基。
还原反应在2-丙醇中进行回流,底物/催化剂比率为1000-10000,加入的碱金属氢氧化物与底物的百分比为2mol%。表1中举例给出各种底物的转化值。值得注意的是,可利用丙酮沸点比2-丙醇低的特点,从反应混合物中分离出2-丙醇氧化生成的丙酮。
还举例给出30℃低压(2-3大气压)条件下,氢气中进行的催化试验,可以看出在这种条件下酮类完全转化为醇类,这表明钌络合物类对于分子氢的氢化反应同样有催化作用。
B1.非手性催化剂的催化试验
所有过程都在氩气中进行,并使用先经脱气的2-丙醇。
实例9:钌(II)络合物类在苯乙酮还原反应中的催化作用。
描述了络合物(10)对苯乙酮还原反应的催化作用。络合物(3-9)的催化试验使用相同方法,结果见表1.
a)络合物(10)催化苯乙酮的还原反应
将络合物(10)(3.5mg,0.005mmol)置入一10ml Shlenk中,加入5ml 2-丙醇制得催化剂溶液。搅拌数分钟,使络合物完全溶解。将苯乙酮(240μL,2mmol)/19ml 2-丙醇溶液置入另一50ml Shlenk中,然后加入1ml配制好催化剂溶液和0.5ml 0.1MNaOH/2-丙醇溶液,反应混合物回流。加入络合物时看做反应开始。苯乙酮/催化剂/NaOH的克分子比率为2000/1/50。
b)用原位制备的络合物(10)催化苯乙酮的还原反应
将络合物(2)(4.3mg,0.005mmol)加入到一10ml Shlenk中,再加入1.0μl2-(H2NCH2)C5H4N(0.01mmol)和5ml 2-丙醇,原位制备催化剂溶液。搅拌数分钟使络合物完全溶解。
将苯乙酮(240μl,2mmol)/19ml 2-丙醇溶液加入另一50ml Shlenk中,再加入1ml配制好的催化剂溶液,和0.5ml的0.1M NaOH/2-丙醇溶液,反应混合物回流。加入络合物时看做反应开始。苯乙酮/催化剂/NaOH的摩尔比率为2000/1/50(表1)。
表1.钌络合物作催化剂的苯乙酮还原为苯基乙醇的反应
[a]碱:K2CO3
实例10:络合物(10)对线状和环状二烷基酮类,烷基芳基酮类和二芳基酮类还原反应的催化作用。
将络合物(10)(3.5mg,0.005mmol)置入一10ml Shlenk中,加入5ml 2-丙醇制得催化剂溶液。搅拌数分钟使络合物完全溶解。
将2mmol酮/19ml 2-丙醇溶液置入另一50ml Shlenk中,加入1ml配制好的催化剂溶液和0.5ml 0.1M NaOH/2-丙醇溶液。加入络合物时看作反应开始。酮/催化剂/NaOH的摩尔比率为2000/1/50。气相色谱分析所得数据见表2。
表2.络合物(10)将酮类催化还原为醇类。酮/络合物/NaOH的摩尔比率为2000/1/50
实验结果表明在络合物(10)的催化作用下,在2-丙醇中回流,线状和环状烷基酮和芳基酮还原为相应的醇类的反应非常迅速,数分钟即可完成,其底物/催化剂比率为2000-5000(见正文)。根据底物的立体的排列和电子的特性不同,转化频率(TOF)值为8000-413000h-1之间(表2)。文献数据表明络合物(10)是活性最强的氢转移催化剂之一,因为除了Mathieu研制的络合物对苯乙酮的TOF值为9000h-1,(H.Yang,M.Alvarez,N.Lugan,R.Mathieu,J.Chem.Soc.,Chem.Commun.1995,1721)以外,其他先前报道的催化剂的TOF值一般均小于10000h-1。
下面举例给出由二苯甲酮合成二苯甲醇的方法,二苯甲醇为一种制备抗组胺及其它药用衍生物的重要的中间体。反应还可使用浓度更高的苯乙酮溶液(1M)作为起始物,然后用蒸馏法去除生成的丙酮。
实例11:二苯基甲醇的合成
在氩气流中,将1.8g二苯甲酮(10mmol)和45ml 2-丙醇置入一100ml烧瓶中,混合物回流。加入2.5ml 0.1M NaOH/2-丙醇溶液和2ml含有催化剂(10)(1.8mg,0.0025mmol)的2-丙醇溶液。二苯甲酮/催化剂/NaOH的摩尔比率为4000/1/100。对反应混合物进行1H核磁共振(NMR)分析,显示30分钟后反应完成。蒸去溶剂后,用30ml二乙醚萃取获得的无色残留物。将该溶剂通过一装有二氧化硅的柱,去除催化剂及氢氧化钠。滤液用Na2SO4脱水,过滤并去除溶剂后,可分离出二苯基甲醇,然后减压干燥(10-2mmHg)。
分离的产量:1.62g(收率:88%)。
因此本方法使用新型催化剂(3-10),以2-丙醇作为氢源可在小至中等规模的醇类合成中,广泛并有效替代化学计量还原剂或分子氢氢化方法。使用催化剂(3-10)可在数分钟内以高还原率将酮定量转化成产品,因此钌络合物是合成多种R2CHOH型醇类及RR’CHOH消旋混合物的理想催化剂,其中RR’CHOH中R,R’基为饱和或不饱和线状或环状脂族,或者芳香羟族-可含有或不含有取代烷基,或含氧,卤素或吡啶原子的取代基。本催化方法具有很高的化学选择性,生成的产品分离简便,可替代目前工业中广泛使用的传统还原剂,如NaBH4,LiAlH4(J.March,Advanced Organic Chemistry,JohnWiley,New York(USA),1984,p.809)以及Al(OC3H7)3(Meerwein-Ponndorf-Verley反应)(H.Meerwein,R.Schmidt,Liebigs Ann.Chem.1925,444,221;A.Verley,Bull.Soc.Fr.1925,37,537;W.Ponndorf,Angew.Chem.1926,39,138;A.L.Wilds,Org.React.1944,2,178)。
B2.手性催化剂的催化试验
所有试验在氩或氢气流中进行,并使用先经脱气的2-丙醇。
实例12:在钌的手性络合物存在下对苯乙酮(0.1M)的光学异构选择性还原。
描述了络合物(12)催化的苯乙酮光学异构选择性还原的方法。用络合物(11-15)同方法,其结果见表3。在氩或氢气流中,用络合物(12)作催化剂的还原反应结果见表4.
a)络合物(12)将苯乙酮光学异构选择性还原为1-苯基乙醇
将手性催化剂(12)(3.6mg,0.005mmol)置入一10ml Shlenk中,加入3ml 2-丙醇制成悬浮液,并加入2ml 0.1M NaOH/2-丙醇溶液,使产物完全溶解。
将苯乙酮(240μl,2mmol)置入另一50ml Shlenk中,加入19ml先经脱气的2-丙醇进行溶解。混合物回流后,加入1ml配制好的催化剂溶液。
苯乙酮/催化剂/NaOH的摩尔比率为2000/1/40。加入络合物时看做反应开始。气相色谱分析所得结果见表3。
表3.在钌的手性络合物存在下将苯乙酮光学异构选择性还原为1-苯基乙醇
反应温度分别为:[a]40℃;[b]70℃;[c]40℃,碱:K2CO3
表4.在30℃下及氩或氢气流中,在络合物(12)存在下,将苯乙酮光学异构选择性还原为1-苯基乙醇
实例13:手性络合物(12)催化酮类的光学异构选择性还原反应
将手性催化剂(12)(3.6mg,0.005mmol)置入一10ml Schlenk瓶中,加入3ml 2-丙醇制成悬浮液,加入2ml 0.1M NaOH/2-丙醇溶液,使产物完全溶解。
将酮(2mmol)置入另一50ml Shlenk瓶中,加入19ml先经脱气的2-丙醇溶解。然后回流,再加入1ml配制好催化剂溶液。
苯乙酮/催化剂/NaOH的克分子比率为2000/1/40.气相色谱分析所得结果见表5。
表5.络合物(12)将酮类光学异构选择性催化还原为醇类。底物/催化剂/碱的摩尔比率为2000/1/40。
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。















动态评分
0.0