

IPC分类号 : B01J13/14,C08G81/00,C08H1/00,A23G4/06,A23L27/00






专利摘要
本发明涉及一种微波合成SF‑Cd缓释微球的制备方法。本发明利用低温脱气处理、降解和纯化分离制得丝素蛋白,并以素蛋白为改性对象,通过微波湿法美拉德反应分别接枝四种糖类物质并筛选,其中环糊精(Cd)的接枝率最高,制得丝素蛋白糖基化接枝产物。本申请的发明人探索不同反应因素对丝素蛋白游离氨基酸含量及褐变程度的影响,同时采用多种表征手段对比了微波加热和水浴加热对SF‑Cd共价复合物的物化性质及结构功能等方面的影响。最后以左旋香芹酮(Left‑Carvon,L‑Ca)为缓释模型物,对丝素蛋白糖基化微球进行口腔模拟缓释性能测试,测定缓释效果。
权利要求
1.一种SF-Cd缓释微球的制备方法,包括如下步骤:
1)低温脱胶
将生蚕茧中的杂质去除并剪碎后,称取一定质量的蚕丝碎片,加入去离子水和无水碳酸钠;使蚕丝完全被溶液浸没,搅拌均匀后密封充入氮气,使之隔绝空气进行脱胶处理,置于37-38℃恒温槽中加入转子匀速搅拌反应;每隔24h取出并过滤,用25%碳酸钠溶液清洗滤出物后,加入等体积的25%碳酸钠溶液继续搅拌反应,每隔24h更换溶液,留取脱胶滤液;
2)脱胶丝素的降解
称取部分冻干后的蚕丝,按照体积质量比丝素蛋白(g):溶剂(mL)=1:70加入溶解丝素蛋白的溶剂;搅拌均匀后密封充入氮气,使之隔绝空气进行降解,置于37-38℃恒温槽中加入转子匀速搅拌反应,三天后取出,在6000r/min条件下低温冷冻离心30min,取上清液;
3)中分子量活性丝素蛋白的分离纯化
将截留分子量分别为20kD和50kD 的透析袋叠加,50kD在内,20kD在外,把上清液倒入50kD透析袋内,在去离子水中透析48h,每隔12h进行换水操作,透析结束后将透析袋间的样品进行冷冻干燥,获得丝素蛋白粉;
4) 多糖的筛选及SF-Cd缓释微球的制备
称取一定质量的丝素蛋白置于烧杯中,再倒入一定量的去离子水,pH值调节至7,配成蛋白含量为10%的溶液;加入多糖,其中质量比为丝素蛋白:多糖=2.5:1,制备出蛋白多糖混合溶液;制备过程中用磁力搅拌装置不停搅拌直至均匀,然后冷冻干燥备用;
配制浓度比的蛋白多糖粉末,将蛋白多糖粉末,研磨均匀后过150目筛,取等量筛出物置于双频超声微波组合催化合成仪中,调节微波功率100w-500w和超声功率100w-300w,反应一段时间后取出,冰水浴1分钟结束反应,即得到SF-Cd混合产物。
2.根据权利要求1所述的制备方法,其特征在于:将蛋白多糖粉末,研磨均匀后过150目筛,取等量筛出物置于双频超声微波组合催化合成仪中,调节微波功率为500w、超声功率为300w、反应时间为4min,反应后取出,冰水浴1分钟结束反应,即得到混合产物。
3.根据权利要求1所述的制备方法,其特征在于:步骤1)中蚕丝质量/g:水体积/L:无水碳酸钠质量/g的比例为10:1:5。
4.根据权利要求1所述的制备方法,其特征在于:步骤2)中溶解丝素蛋白的溶剂为含20%乙醇及40%氯化钙的溶液;含30%乙醇及40%氯化钙的溶液,或含40%乙醇及40%氯化钙的溶液。
5.根据权利要求1所述的制备方法,其特征在于:步骤2)中溶解丝素蛋白的溶剂为含30%乙醇及40%氯化钙的溶液。
6.根据权利要求1所述的制备方法,其特征在于:步骤4)中多糖为环糊精、淀粉、麦芽糊精、琼脂糖。
说明书
技术领域
本发明涉及一种微波合成SF-Cd缓释微球的制备方法。
背景技术
口香糖是一种由天然树胶、糖浆和香精香料经一定的加工工艺制备而成的休闲糖果,质量好的口香糖往往咀嚼时间较长,这就要求其使用的香精香料在口中耐咀嚼并保持释放时间持久。因此,选择高浓度、溶解性较小的香精是口香糖制备过程的重要环节。目前应用于食品的固体香精大都没有缓释性能或者缓释能力较差。由于在生产能力和成本上具有优势,现有的大部分食品香精制备大多采用喷雾干燥包埋法
丝素蛋白(Silk Fibroin,SF)因其良好的生物相容性和缓释性能,被广泛应用与医药食品行业,同时,由于其降解效能良好,不会对环境造成污染,是一种高效、应用范围广、可持续的生物材料。然而仅以丝素蛋白作为缓释微球,其亲水性较差,且在缓释初期容易爆释负载物,会导致释放速率的突跃变化影响缓释效果。有研究发现,微波美拉德(Maillard Reaction by
目前蚕丝脱胶使用较多的方法有高温高压沸水脱胶、尿素脱胶、酶脱胶和碳酸钠脱胶等等。不同的脱胶方法会影响产物的溶解性、后期的水解效率、分子量等。尤其是高温高压处理法对丝素的拉伸率和断裂力度影响较大,均有下降,碱性蛋白酶法脱胶后产物的白度提高明显。
脱胶后的丝素不溶于水,经过盐溶液或有机溶剂水解后方可得到丝素蛋白溶液。目前,应用较多的是溴化锂或三元体系降解液来对丝素进行水解。Park等
发明内容
本发明的目的在于提供一种乳化性能和起泡性能较好的SF-Cd缓释微球的制备方法。
一种SF-Cd缓释微球的制备方法,包括如下步骤:
1)低温脱胶
将生蚕茧中的杂质去除并剪碎后,称取一定质量的蚕丝碎片,加入去离子水和无水碳酸钠;使蚕丝完全被溶液浸没,搅拌均匀后密封充入氮气,使之隔绝空气进行脱胶处理,置于37-38℃恒温槽中加入转子匀速搅拌反应;每隔24h取出并过滤,用25%碳酸钠溶液清洗滤出物后,加入等体积的25%碳酸钠溶液继续搅拌反应,每隔24h更换溶液,留取脱胶滤液;
2)脱胶丝素的降解
称取部分冻干后的蚕丝,按照体积质量比丝素蛋白(g):溶剂(mL)=1:70加入溶解丝素蛋白的溶剂;搅拌均匀后密封充入氮气,使之隔绝空气进行降解,置于37-38℃恒温槽中加入转子匀速搅拌反应,三天后取出,在6000r/min条件下低温冷冻离心30min,取上清液;
3)中分子量活性丝素蛋白的分离纯化
将截留分子量分别为20kD和50kD的透析袋叠加,50kD在内,20kD在外,把上清液倒入50kD透析袋内,在去离子水中透析48h,每隔12h进行换水操作,透析结束后将透析袋间的样品进行冷冻干燥,获得丝素蛋白粉;
4)多糖的筛选及SF-Cd缓释微球的制备
称取一定质量的丝素蛋白置于烧杯中,再倒入一定量的去离子水,pH值调节至7,配成蛋白含量为10%的溶液。加入多糖,其中质量比为丝素蛋白:多糖=2.5:1,制备出蛋白多糖混合溶液。制备过程中用磁力搅拌装置不停搅拌直至均匀,然后冷冻干燥备用;
将上述粉末,研磨均匀后过150目筛,取等量筛出物置于双频超声微波组合催化合成仪中,调节微波功率为500w、超声功率为300w、反应时间为4min,反应后取出,冰水浴1分钟结束反应,即得到混合产物;
配制浓度比的蛋白多糖粉末,以同样方法处理并置于双频超声微波组合催化合成仪中反应,调节微波功率功率和超声功率,反应一段时间后取出,冰水浴1分钟结束反应,即得到SF-Cd混合产物。
其中,
步骤1)中蚕丝质量/g:水体积/L:无水碳酸钠质量/g的比例为10:1:5。
步骤2)中溶解丝素蛋白的溶剂为含20%乙醇及40%氯化钙的溶液;含30%乙醇及40%氯化钙的溶液,或含40%乙醇及40%氯化钙的溶液。
步骤2)中溶解丝素蛋白的溶剂为含30%乙醇及40%氯化钙的溶液。
步骤4)中多糖为环糊精、淀粉、麦芽糊精、琼脂糖。
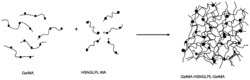
本章的目的是以丝素蛋白为改性对象,通过Maillard反应接枝β-Cd,制成SF-Cd缓释微球来克服丝素蛋白的初期爆释问题,同时β-Cd本身也具有一定的包载性能,可使微球的包载率增加。由于丝素蛋白的空间结构多为β-折叠,β-Cd为多糖结构,其反应位点较少,从而导致Maillard反应时间较长。为解决此问题本课题选择了微波Maillard法来提高反应速率。
其中,本发明利用低温脱气处理、降解和纯化分离制得丝素蛋白,并以素蛋白为改性对象,通过微波湿法美拉德反应分别接枝四种糖类物质并筛选,其中环糊精(Cd)的接枝率最高,制得丝素蛋白糖基化接枝产物。
本申请的发明人探索不同反应因素对丝素蛋白游离氨基酸含量及褐变程度的影响,同时采用多种表征手段对比了微波加热和水浴加热对SF-Cd共价复合物的物化性质及结构功能等方面的影响。最后以左旋香芹酮(Left-Carvon,L-Ca)为缓释模型物,对丝素蛋白糖基化微球进行口腔模拟缓释性能测试,测定缓释效果。主要研究内容和结论如下:
1、选用较温和的低温脱气处理,极大保留丝素蛋白的生物活性。经低温脱胶、降解,再利用创新重叠透析法将目标分子量丝素蛋白纯化分离,此时目标产物可溶性丝素蛋白制得率为24.23%,经常规成分分析可得,实验室自制中分子量可溶丝素蛋白的纯度较高,易于后期改性。研究丝素蛋白乳化性能和起泡性能,在低浓度时,乳化性能随丝素蛋白的浓度升高而增加,且浓度越高,乳化稳定性越好,同时pH值为8时的乳化性能相对较高,不同pH对丝素蛋白乳化稳定性影响不大;当浓度为1%时,起泡能力最强,浓度高于0.5%时,起泡稳定性较好。利用FTIR、DSC、SEM等表征手段对产物结构进行分析。
2、以丝素蛋白为改性对象,通过湿法美拉德反应分别接枝β环糊精、淀粉、麦芽糊精和琼脂糖,选择微波美拉德法来提高反应速率,制得丝素蛋白糖基化接枝产物,根据接枝率筛选出丝素蛋白接枝环糊精为最优产物,探究其超声功率,反应时间,微波功率,底物配比等因素对丝素蛋白游离氨基酸含量及褐变程度的影响,同时经响应面分析法最后得出丝素蛋白糖基化最佳反应条件为:底物配比丝素蛋白:β-Cd=2.5:1、微波功率为500w、反应时间为4min,通过实验验证所得接枝率为82.95%,此时得到的丝素缓释蛋白性能优异。
3、探究微波前后的溶解性、白度、二硫键和巯基等含量的变化,并采用多种表征手段探讨微波对SF-Cd共价复合物的物化性质及结构功能等方面的影响。结果表明,经过微波后的产物溶解性相较微波前有较大差异。丝素蛋白原料和水浴加热后的SF+Cd在pH为4的条件下溶解性最低,而微波后的SF+Cd在pH为6的条件下溶解性最低;荧光光谱图显示,在激发波长360nm的条件下,丝素蛋白与β-Cd微波糖基化产物发射波长的最强荧光强度出现在429nm处,微波糖基化后的丝素蛋白荧光强度明显高于水浴加热和未经处理的丝素蛋白,在反应3min时,荧光强度明显增强,表明3-4min时的产物生成速率加快,美拉德反应更强烈;FT-IR图谱表明,丝素蛋白与环糊精通过碳氨缩合反应生成糖基化产物;DSC实验结果说明产物热稳定性良好;SEM图像观察可知丝素蛋白经糖基化后表面粗糙度增加,有利于后期的吸附应用。
4、以香芹酮为缓释模型物,对SF-Cd微球进行口腔模拟缓释性能测试,并探究防腐剂、pH等因素对其缓释效果的影响。结果表明,负载香芹酮的微球的负载率8.27%、包封率80.65%,在口腔pH及温度条件下的缓释时间可达30min,缓释速率适宜;食品添加剂对产物缓释效果影响较小,实际应用中可忽略。探究丝素蛋白糖基化微球的缓释机理发现,该释放过程符合Fickian扩散,且生物降解性良好。
附图说明
图1时间对脱胶效果的影响。
图2不同水解条件下的产物的SDS-PAGE。
图3浓度对丝素蛋白乳化性能的影响。
图4溶液pH对丝素蛋白乳化性能的影响。
图5浓度对丝素蛋白乳化稳定性的影响。
图6溶液pH对丝素蛋白乳化稳定性的影响。
图7丝素蛋白浓度对起泡能力及起泡稳定性的影响。
图8丝素蛋白的红外光谱图。
图9丝素蛋白的DSC热分析图。
图10丝素蛋白的电镜图。
图11不同超声功率与时间对接枝率的影响。
图12不同超声功率与时间对褐变程度的影响。
图13微波功率对接枝率和褐变程度的影响。
图14底物浓度比对接枝率和褐变程度的影响。
图15底物配比、微波功率和微波时间对接枝率影响的响应面分析图。
图16微波加热对丝素蛋白糖基化中间产物的影响。
图17溶液pH对产物Zeta电位及粒径大小的影响。
图18不同加热方式对产物二硫键含量的影响。
图19不同加热方式对产物巯基含量的影响。
图20不同加热方式对产物溶解性的影响。
图21不同加热条件下产物的荧光光谱图。
图22不同加热方式产物的红外光谱图。
图23不同加热方式下的DSC热分析图。
图24不同加热方式下的产物电镜图(1)环糊精(2)丝素蛋白(3)水浴加热产物(4)微波加热产物。
图25不同香芹酮浓度对微球包封率和负载率的影响。
图26不同pH条件下的产物微球的体外缓释性能。
图27不同温度条件下的产物微球的体外缓释性能。
图28微波对产物缓释性能的影响。
图29食品添加剂对产物缓释性能的影响。
具体实施方式
实施例1丝素蛋白粉的制备方法
用于制作SF-Cd缓释微球的丝素蛋白粉的制备方法,包括如下步骤:
1)低温脱胶
传统的脱胶方式较剧烈,需在高温条件下煮沸数小时,且要进行强酸强碱处理,这些方法会一定程度上破坏丝素蛋白的空间结构从而导致后续应用性能的降低,甚至使其丧失活性。本课题选用较温和的低温脱气处理,将极大保留丝素蛋白的生物活性。
将生蚕茧中的杂质去除并剪碎后,称取一定质量的蚕丝碎片,按照比例(蚕丝质量/g:水体积/L:无水碳酸钠质量/g=10:1:5)加入去离子水和无水碳酸钠。使蚕丝完全被溶液浸没,搅拌均匀后密封充入氮气,使之隔绝空气进行脱胶处理,置于37-38℃恒温槽中加入转子匀速搅拌反应。每隔24h取出并过滤,用25%碳酸钠溶液清洗滤出物后,加入等体积的25%碳酸钠溶液继续搅拌反应,每隔24h更换溶液,留取脱胶滤液,进行紫外光谱测定。
脱胶结束后,取适量蚕茧碎片放到小烧杯中,用1%的苦味酸胭脂红溶液检测脱胶效果,使样品浸没于苦味酸胭脂红溶液中,煮沸5min后用去离子水冲洗并观察蚕丝颜色
2)脱胶丝素的降解
称取部分冻干后的蚕丝,分成六等份。按照体积质量比丝素蛋白(g):溶剂(mL)=1:70加入下列溶解丝素蛋白的溶剂:①40%的氯化钙溶液;②60%的氯化钙溶液;③含20%乙醇及40%氯化钙的溶液;④含30%乙醇及40%氯化钙的溶液;⑤含40%乙醇及40%氯化钙的溶液;⑥60%的乙醇溶液。搅拌均匀后密封充入氮气,使之隔绝空气进行降解,置于37-38℃恒温槽中加入转子匀速搅拌反应,三天后取出,在6000r/min条件下低温冷冻离心30min,取上清液。
3)中分子量活性丝素蛋白的分离纯化
初步分离出的丝素蛋白分子量范围较大,分子内部折叠复杂,不利于后期改性
将截留分子量分别为20kD和50kD的透析袋叠加,50kD在内,20kD在外,把上清液倒入50kD透析袋内,在去离子水中透析48h,每隔12h进行换水操作,此方法不仅可以去除分子量大于50kD的基本未降解的蚕丝,同时可以滤出分子量小于20kD的降解过于充分的丝素蛋白以及醇、盐类,防止蛋白溶液自发凝胶。透析结束后将透析袋间的样品进行冷冻干燥,获得丝素蛋白粉。
实施例2理化指标分析及表征
(1)常规成分的测定
蛋白质的测定:微量凯氏定氮法(GB5511-85)
脂肪的测定:索氏抽提法(GB5497-85)
水分的测定:105℃恒重法(GB5512-85)
灰分的测定:干法灰化法(GB5505-85)
(2)乳化性及乳化稳定性的测定
经氯化钙降解后,丝素蛋白会被赋予良好的乳化性、起泡性及稳定性,这些良好的理化性质决定着丝素蛋白在医疗食品等领域的应用
分别取一定量的丝素蛋白,配成浓度为0.25%、0.5%、1%、2%浓度的丝素蛋白溶液,加入缓冲溶液调节pH值为3、5、8、10。分别加入等体积的色拉油,分散5min在10000r/min转速下,取10mL离心5min,取出后离心管中的乳业分为三层,从上至下分别为油层、乳化层和水层,乳化能力通过测出油层的高度用以下公式计算得:
按以上方法取样配制,分散1min在10000r/min转速下,用量筒取10mL存放在25℃条件下,24h后取出,测定油层、乳化层和水层的高度。乳化稳定性通过以下公式计算得:
(3)起泡性及起泡稳定性的测定
取上述浓度为0.25%、0.5%、1%、2%的丝素蛋白溶液10mL,分散于转速为10000r/min的条件下2min,停止后立即测定泡沫高度。
转动停止30min后,测量泡沫的高度。
(4)紫外分光光度法
对上述实施例1中步骤1)中的脱胶溶液稀释后进行紫外光谱测定
(5)红外光谱图
准确称取样品适量,以质量比为1:50的量加入一定量的溴化钾,用研钵研磨至样品和溴化钾充分混合成均匀的粉末,在压片机上将混合粉末压成薄片,再用傅立叶红外分光光度计作全波段(4000-400cm
(6)差热DSC扫描
取3-8mg左右冷冻干燥后的丝素蛋白产物,记下准确质量,放入铝盘中,将样品平铺于铝盘中并用盖子压实,设置温度扫描范围为40-350℃,升温程序:20℃/min。载气N2。载气流量:20mL/min。每次实验前先扫描空吕盘作为空白对照。
(7)扫描电子显微镜分析
取一定量的待测样品,粘在实现剪裁好的导电胶上,用离子溅射镀膜仪在样品表面进行喷金,镀完金膜后将样品放入电子显微镜的放样室中推入观察。
(8)聚丙烯凝胶电泳
根据文献,作以下处理:
将丝素蛋白产物配成蛋白浓度为1mg/mL的溶液,取一定量与SDSβ-巯基乙醇以1:2体积比混合,煮沸后进样,进样量为10L。分离胶和浓缩胶的浓度分别为12%和5%,电流为15mA,等到样品流至分离胶后调节电流至20mA,当距离底边还有1cm时,电泳停止。用考马斯亮蓝进行染色后用脱色液脱色,然后用蒸馏水浸泡过夜。电泳胶的配置方法如表2-3:
表2-3凝胶电泳组成成分表
Table 2-3 Composition of gel electrophoresis
结果与讨论
1低温脱胶效果
将每次过滤保留的溶液稀释十倍后测定其在280nm处的吸光度,如图1所示。根据图中所示可以看出第二天的脱胶滤液中的丝胶蛋白含量最高。丝胶蛋白含量最高点在第二天出现,主要是由于第一天碳酸钠溶液对蚕丝溶胀不彻底。蚕茧中的丝胶蛋白与丝素蛋白结合紧密,层层粘连,形成不规则的交叉网状结构,导致不易迅速溶胀完全。继续对蚕丝进行溶胀后,如图所示,脱胶液在第四天吸光值接近0,并且在第五天趋于稳定。同时,对脱胶后的蚕丝产物进行苦味酸胭脂红溶液检测,颜色结果呈黄色,由此也可推出产物已脱胶完全。且此时的脱胶丝素蛋白产率为67.21%,与丝素蛋白理论含量70%接近。脱胶后的丝素蛋白经过冷冻干燥后颜色白皙,质感较好。
2脱胶丝素降解
丝素蛋白主要由18种氨基酸中的三种氨基酸组成,分别是甘氨酸、丙氨酸、丝氨酸,这三种氨基酸占到总组分的80%以上。蚕丝在生物体内产生初期,以可溶性氨基酸无规则排列卷曲而成,无明显结构特征且基本无活性,当进一步卷曲形成较高级结构时,丝素分子会通过自组装形成线形聚集体,此时由于β折叠方式的出现和其他高级构象的形成,丝素的抗物理剪切能力和化学稳定性明显加强。有研究表明,氯化钙可以有效地降解丝素纤维。其机理是,当把处于稳定状态的的丝素浸没到一定浓度的氯化钙溶液中时,此时大量的强极性离子会产生强水化作用,使丝素表面附着大量的水,水会显著增强丝素中多肽链的运动,从而丝素分子间的范德华力及氨基酸残基侧链的氢键被破坏。同时,氯化钙会和丝素分子中的极性氨基酸直接发生反应,如丝素中大量存在的酪氨酸,且酪氨酸为暴露型氨基酸残基,氯化钙易于与之发生反应,由于在丝素中的结晶区与非结晶区均存在大量酪氨酸,故这一反应会极大破坏丝素结构及性质
经过六组不同配比的降解溶液的反应后,观察结果后发现,与用蒸馏水作参照的空白实验相比,六组中的⑥(60%的乙醇溶液)与空白实验一样,丝素没有降解;①和②中的丝素也只有少量的降解;在以三元体系为降解体系的③④⑤中,丝素均有降解且速度快降解量多,其中以④(含30%乙醇及40%氯化钙的溶液)的降解量最多且速度最快,故选择含30%乙醇及40%氯化钙的溶液作为丝素的三元降解体系。
如图2,通过聚丙酰胺凝胶电泳进一步论证,在③④⑤条件下分别水解1、2、3d,结果如下:从图中可以看出,在④含30%乙醇及40%氯化钙的溶液中条件下的丝素水解速度最快,且水解程度彻底,其水解产物的水溶性较其他两组好;在20-50kD分子量范围的产物较多。
3可溶性活性丝素蛋白的制备
降解所得的丝素蛋白分子量区间较大,其中分子量小于10kD的丝素肽段缓释活性较差,分子量大于50kD的丝素蛋白基本未降解且不易于改性,故目标丝素蛋白即为分子量20-50kD。基于此,选择该分子截留区段的两种透析袋,透析结束后两层透析袋之间的即为目标丝素蛋白。
4目标丝素蛋白的得率与提取率
脱胶产率
即为脱胶后丝素质量占原蚕丝质量的比值,计算得为T=67.21%。
降解丝素蛋白得率
式中:G:降解丝素蛋白得率
Mr:脱胶后的丝素质量
Ms:降解所得丝素蛋白的质量
计算得为96.15%
目标丝素蛋白提取率
式中:K:目标可溶丝素蛋白的提取率
Mm:目标可溶丝素蛋白的质量
Ms:降解所得丝素蛋白的质量
计算得为37.50%,即目标可溶性丝素蛋白制得率为P=T*G*K=24.23%。
5理化指标分析及表征
常规成分的测定
本研究课题中,实验室自制的丝素蛋白中的蛋白质含量为92.96%,脂肪含量为0.57%,水分含量为4.42%,灰分2.34%,见表2-4:
表2-4丝素蛋白的主要成分
Table 2-4 The main composition of SF
从表2-4中可看出,实验室自制的丝素蛋白的蛋白含量较高,纯度较高,可以用来进行实验研究。
乳化性及乳化稳定性的测定
乳化性能指的是每克蛋白所能乳化油的体积
如图4所示,在不同的pH条件下的丝素蛋白乳化性能变化不明显,当pH值为8时的乳化性能相对较高,这主要是由于此时的溶液pH接近丝素原溶液的pH,有较好的相容性,从而使丝素蛋白在较大的pH范围内均有良好的乳化性能。
从图5中可以看出,在低浓度时,浓度越高,乳化稳定性越好,当浓度高于0.5g/mL时,乳化稳定性提高得不明显,这是由于当浓度达到一定时,经丝素蛋白乳化后的液体是一个高度分散体系,有着极大的表面势能,此时水油界面的的表面张力减小,乳化后的界面会产生膜效应,有较大的机械强度,使体系的稳定性提高,膜效应随着乳化剂浓度的提高而提高。当弱酸性的油液与中性的丝素蛋白溶液相溶时,此时的溶液pH值接近丝素蛋白的等电点,易发生凝胶现象。
如图6所示,在一定浓度下(0.5%),不同pH对丝素蛋白乳化稳定性影响不大,在pH为10时才略有下降。蛋白质的乳化稳定性与高速剪切过后形成的界膜稳定性有关,丝素蛋白所形成的界膜对溶液pH值变化不敏感,所以导致其包覆的油在界膜中不易溢出,从而乳化性稳定。
起泡性及起泡稳定性的测定
如图7所示,丝素蛋白浓度对起泡能力的影响,随着浓度的升高,起泡能力明显增强,当浓度高于1%时丝素蛋白起泡能力下降,这主要是由于高浓度的丝素蛋白溶液中含β-折叠结构。与其他蛋白相比,在同条件下的丝素蛋白的起泡能力略低于酪蛋白,这是由于丝素分子中所含的疏水基团较酪蛋白少,疏水基团的多少可以有效影响蛋白的起泡能力。
丝素蛋白浓度对起泡稳定性的影响如图所示,在低浓度时,起泡稳定性较差,当浓度高于0.5%时,起泡稳定性较好且趋于平稳,这是由于在高浓度下的蛋白仅部分展开折叠结构,此时在形成的界面的解吸速率慢,与低浓度的蛋白相比,它能形成更加稳定的泡沫结构。具有折叠结构的蛋白会形成环状非共价结构,此结构具有强相互作用且会延伸至水相,促进了蛋白形成稳定的网状结构。
红外光谱图
红外结果表明,在1650cm
差热DSC扫描
丝素蛋白的DSC热分析图谱如图9所示。可以看出,丝素蛋白的热反应集中于180-185℃这一小范围内,在具体应用中可体现出较优的热稳定性。从丝素蛋白的一级结构分析,它由18种氨基酸组成,在这18种氨基酸中,Thr,Ser,Arg,Pro为最不稳定氨基酸,His和Asn则为次不稳定氨基酸,不稳定氨基酸越多则丝素蛋白越不稳定
扫描电子显微镜分析
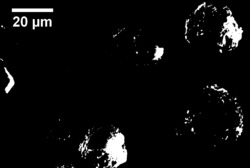
拍摄400和2000倍丝素蛋白电镜图,如10所示,可以看出丝素蛋白呈球状且颗粒较为分明,颗粒间无粘连无团聚。
结论:
(1)本申请选用较温和的低温脱气处理,极大保留丝素蛋白的生物活性。经低温脱胶,脱胶后丝素降解,再利用创新重叠透析法将目标分子量丝素蛋白纯化分离,此时目标可溶性丝素蛋白制得率为24.23%,经常规成分分析可得,实验室自制中分子量可溶丝素蛋白的纯度较高,易于后期改性。
(2)研究丝素蛋白乳化性能和起泡性能。在低浓度时,乳化性能随丝素蛋白的浓度升高而增加,且浓度越高,乳化稳定性越好,同时pH值为8时的乳化性能相对较高,不同pH对丝素蛋白乳化稳定性影响不大;当浓度1%时,起泡能力最强,浓度高于0.5%时,起泡稳定性较好。
(3)经多种表征手段结果表明,在实验室自制的目标可溶丝素蛋白中:同时存在相对稳定的α-螺旋结构和β-折叠结构;有较好的热稳定性;颗粒表面光滑规则无团聚。
4)微波合成
实验材料
表3-1主要实验材料
Table 3-1 Main experiment materials
2实验方法
2.1多糖的筛选及SF-Cd缓释微球的制备
称取一定质量的丝素蛋白置于烧杯中,再倒入一定量的去离子水,pH值调节至7,配成蛋白含量为10%的溶液。加入不同多糖(环糊精、淀粉、麦芽糊精、琼脂糖),其中质量比为丝素蛋白:多糖=2.5:1,制备出蛋白多糖混合溶液。制备过程中用磁力搅拌装置不停搅拌直至均匀,然后冷冻干燥备用。
将上述四种不同粉末,研磨均匀后过150目筛,取等量筛出物置于双频超声微波组合催化合成仪中,调节微波功率为500w、超声功率为300w、反应时间为4min,反应后取出,冰水浴1分钟结束反应,即得到混合产物。根据2.2方法测定四种产物中游离氨基酸量,计算得到接枝率,选择接枝率最高的蛋白多糖组合继续实验。
配制不同底物浓度比的蛋白多糖粉末,以同样方法处理并置于双频超声微波组合催化合成仪中反应,调节微波功率功率(100w、300w、500w、700w、900w)和超声功率(100w、200w、300w),反应一段时间后取出,冰水浴1分钟结束反应,即得到SF-Cd混合产物。
2.2游离氨基的测定接枝率的计算
蛋白质与糖类主要通过蛋白中的游离氨基与糖类的羧基之间的反应,游离氨基数量越少,接枝率越高,反应程度也就越高,因此,可以通过测得的游离氨基酸数量来计算接枝率。
本文采用OPA法
准备两种试剂,分别为CA试剂和CB试剂。
CA试剂:取0.04g的邻苯二甲醛(OPA),加入1mL甲醇和3mL去离子水溶解,保存于棕色试剂瓶中备用。
CB试剂:将2.5mL 20%的十二烷基磺酸钠(SDS),25mL 0.1mol/L的硼砂,0.1mL的β-巯基乙醇混合加入50mL容量瓶后定容备用。
取配好的上述溶液CA试剂0.3mL和CB试剂7mL置于试管中混合均匀,并加入0.2mL的含0.01g/mL丝素蛋白的样品液混匀,在35℃条件下反应2min,测定340nm处吸光值,同时做空白溶液(以等量水代替样品加入),测得的两个数值之差即为游离氨基净吸光值。根据标准曲线(赖氨酸)和净吸光值计算游离氨基酸含量C,并计算接枝率,来判定反应程度。计算公式如下:
式中:DG:接枝率
C0:反应前的游离氨基含量
Ct:反应t时的游离氨基含量
2.3反应物褐变程度的测定
随着糖基化反应的进行,会有类黑素的产生,并且随反应时间的增加而加深。现取反应冷却后所得的丝素蛋白-多糖样品,研磨至粉碎,放入冷冻离心机4000r/min离心5min,取出样品用质量浓度为0.1%的十二烷基磺酸钠(SDS)溶液稀释20倍,以SDS溶液为空白对照,测定其在420nm处的吸光值。用吸光值表示褐变程度,吸光值越大,褐变程度越高,反应越彻底。
2.4中间产物的测定
根据高威
2.5白度的测量
采用上海汉普光电科技有限公司生产的便携式色差仪HP-2132对油炸甘薯片进行测定,得出L、a、b三个指标,L代表明度[0,100],a代表红/绿差异[127,-128],b代表黄/蓝差异[127,-128]。样品色泽的优劣以L值和b值作为判断标准。每个样品测定10次,取平均值,通过亨特(Hunter)白度公式计算白度值。该公式将完全反射漫射体的白度定义为100,把样品的白度与完全反射漫射体的白度进行对比,以计算色差的方式来评价样品的白度。
式中,K1是常数,一般情况取值为1。ap,bP是理想白在Lab系统中的白度指数,一般情况下:
测量不带荧光的样品时ap=0.00,bP=0.00;
测量带有荧光的样品时ap=50,bP=-15.87:
2.6粒径大小及分布分析
将不同制备条件下的丝素蛋白糖基化产物加入比色皿中,在25℃条件下用粒度分析仪检测粒径大小及分布,同时测量Zeta电位。
3结果与讨论
1微波场中各参数对产物影响
1.1超声功率与时间
研究表明,Maillard反应速率随反应超声功率的升高而加快,当超声功率每升高50w,反应速率会提升1-2倍,这是由于超声功率的升高使蛋白和多糖震动加剧,从而导致结构展开,暴露出更多的反应活性基团,从而使Maillard反应的活性位点(羰基和氨基)增加,反应加速,接枝率升高
超声功率与反应时间对蛋白质糖基化反应有着很大的影响,主要影响反应进程的中间产物及终产物。在本实验中,我们控制反应微波功率400w,设定功率恒定,相对湿度为79%,反应底物浓度比为丝素蛋白:环糊精=2.5:1;同时控制超声功率与时间变量,超声功率(100w、200w、300w),时间(1min、2min、3min、4min、5min、6min、7min),研究不同超声功率与反应时间条件下的糖基化接枝率与褐变程度的变化,以选取最优条件,具体结果如图。
从图11中我们可以看出,不同超声功率与反应时间对接枝率的影响。随着微波时间的增加,在2-16h时,接枝率随微波时间增加而上升较快,300w时接枝率从1min的51%提升到了4min时的86.18%;而随着微波时间的继续增加,接枝率出现缓慢下降,最后趋于平衡,在7min接枝率为81.95%。这可能是由于在微波时间低于4min时,丝素蛋白的活性位点随时间的增加而逐渐打开发生接枝化反应,而当时间达到4min时,反应基本完成,活性位点基本反应完全,且反应的延长使得反应物和反应产物发生一定程度的水解
从超声功率来看,反应时间为4min时,反应在100w、200w、300w三个功率下进行,接枝率分别为69.45%、830%、86.18%。由此可见在300w时接枝率最高,这可能是由于在100w-200w时,丝素蛋白与多糖结构蜷缩,反应结构不易暴露,超声功率升高对反应影响较大,有助于打开活性位点,而当功率增加到一定时,影响减小但仍有影响。
从图12中我们可以看出,不同超声功率与反应时间对褐变程度的影响。Maillard反应所产生的褐变物质会干扰反应产物的纯度与色泽,影响后期应用。当反应时间为4min时,比较不同超声功率对褐变程度的影响。可以看出在200w时的褐变指数相对较低,为0.144。结合图11,寻找合适条件保证糖基化产物在褐变程度较小的情况下达到可观的接枝度,故确定微波时间为4min,超声功率为200w,此时的反应产物效果可以达到相对最佳。
1.2微波功率
反应微波功率对蛋白质糖基化反应有着很大的影响,主要影响反应进程的中间产物及终产物。在本实验中,我们控制超声功率为200w,反应时间为4min,编辑功率恒定,相对湿度为79%,反应底物浓度比为丝素蛋白:环糊精=2.5:1;同时控制微波功率变量(100w、300w、500w、700w、900w),研究不同微波功率条件下的糖基化接枝率与褐变程度的变化,以选取最优条件,具体结果如下图13。
从图13中我们可以看出微波功率对接枝度和褐变程度的影响。由图可知,在100w-500w时,接枝率随功率的增加而明显增加,当功率高于500w时,接枝率随功率增加而增速放缓。这是由于功率较低时升高功率有利于反应体系的升温,反应位点打开,反应速率升高,而当功率超过500w时,由于微波场内丝素蛋白分子间发生了物理团聚
1.3底物配比
底物浓度的配比对糖基化反应的速率与进程有着很大的影响,在接枝率与褐变程度上均有反映。在本试验中,我们控制超声功率为200w,反应时间为4min,微波功率500w,编辑温度恒定,相对湿度为79%,初步选定底物配比浓度为丝素蛋白:环糊精(1:1,1.5:1,2:1,2.5:1,3:1),研究不同底物浓度配比条件下的糖基化接枝率与褐变程度的变化,以选取最优条件,具体结果如下图。
由图14我们可以看出,褐变程度随底物配比浓度(丝素蛋白:环糊精)的增加而持续增加,且类黑素的积累速度加快,而就接枝度来看,随着底物配比浓度的增加,在2.5:1时达到顶峰,之后缓慢下降,由图我们选择2.5:1的底物配比浓度作为实验条件。
2接枝反应条件的优化
2.1模型回归方程分析
结合单因素实验所筛选的实验条件,为了验证结论的正确性,通过响应面软件Design-Expert 9用来进行数据的多元回归分析。根据单因素实验结果,选择微波反应温度、时间、功率为影响因素,接枝率作为响应值。利用Box-Benhnken模型设计实验,我们得到了独立变量:A(微波功率)、B(底物配比)、C(微波时间),响应面实验结果及模型回归方程方差分析如下:
表3-3响应面实验结果
Table 3-3 The results of the Response Sursace Design
表3-4模型回归方程方差分析
Table 3-4 ANOVA for Response Surface Quadratic Model Analysis ofvariance table
用响应面程序对接枝率Y(%)响应值进行回归分析,最后得出回归方程如下:
Y=81.86-0.066A-33B-0.24C+0.077AB+0.88AC+1.34BC
-11.46A
从模型回归方程方差分析表3-4中我们可以看出,各单因素对响应值的影响中,底物配比和微波时间不显著,非线性关系,微波功率P值小于0.05,对响应值影响较大;三个单因素两两交互对响应值的影响不显著,说明各单因素的交互作用对响应值变化影响较小;模型回归方程的P值小于0.05,说明方程拟合度良好;同时可以看出各二次项P值均小于0.05,说明各单因素的二次项对实验结果影响较大;同时,回归模型的相关系数R
2.2响应面直观分析
通过响应面可视分析软件Design-Expert 9探究变量之间的交互影响以确定最佳反应条件。结果如图15所示。响应面图可以直观地反映各因素对响应值Y(接枝率)的交互影响。当响应曲面的坡度较缓,斜率较小时,说明该变量对响应值影响不大;而当响应曲面的坡度较陡峭是,说明该变量对响应值影响大,调整该变量时响应值将受较大影响。
结合回归方程和响应面分析,优化后的结果表明:(a)底物配比与微波功率对接枝率的影响,随微波功率的增加而升高,底物配比为2.5:1时的接枝率最高。(b)底物配比与微波时间对接枝率的影响,在4min、2.5:1时接枝率达到最大。(c)微波功率与微波时间对接枝率的影响,随微波功率的增加而升高,微波时间为4min时的接枝率最高。
2.3反应最佳优化条件
经过模型拟合所得的理论最佳条件为底物配比丝素蛋白:β-Cd=2.5:1、微波功率为500w、反应时间为4min,在此最佳条件下预计接枝率为807%。通过实验验证这一理论值,所得接枝率为82.95%,理论值与实际值的相对误差较小,表明该优化工艺参数的方程可以指导实际合成。
表3-5理论与实验对比
Table 3-5 The contrast of the experimental value and the theoreticalvalue
3中间产物
为了检测Maillard反应的中间产物,一般测定294nm处的吸光值
4白度的测量
样品白度的优劣以L值、a值和b值作为判断标准。每个样品测定10次,得到L、a、b值后通过亨特(Hunter)白度公式计算白度值W
表3-6糖基化丝素蛋白白度值的计算
从表3-6中计算白度平均值为76.94。白度值W是判断糖基化丝素蛋白性状的重要指标之一,W值越接近100越白
5pH对产物Zeta电位及粒径大小的影响
该实验测定了不同的环境pH值下的微波糖基化丝素蛋白表面的Zeta电位及粒径变化,有助于指导产物在不同环境中的应用情况,结果如图17所示。结果表明,微波糖基化丝素蛋白表面Zeta电位从pH为3时的22mV变化到pH为9时的-35.7mV,呈单调递减趋势,电势0点出现在pH5-6之间。当pH值在7-9时,其电位分别为-19.3mV、-29.5mV、-35.7mV。此时的pH远离等点带,颗粒带较强负电,且糖基化丝素蛋白颗粒间的静电排斥力,使颗粒间相互挤压粒径较小,此时无凝集产生。pH为5和6时的电位分别为4.2mV和-13mV,此时粒径分别为1.81μm和1.69μm,明显高于其他pH值下的粒径大小,出现明显凝集。这是由于此时溶液接近等电点,粒子间作用力以范德华力为主。当pH值为3和4时,微波糖基化丝素蛋白表面Zeta电位分别为22.8mV和15.3mV,几乎无絮凝产生,此时的粒子绝对电位值与pH为8时接近,但粒径却远大于pH为8时的粒子直径,这是由于溶液pH调节以pH=7为起点,当调节到pH3-4时经历了pH为5左右时的不可逆絮凝,导致pH3-4时的粒径大于理论值。
4本章小结
(1)以丝素蛋白为改性对象,通过Maillard反应接枝β-Cd,选择了微波Maillard法来提高反应速率,制得丝素蛋白糖基化接枝产物。探究了超声功率,反应时间,微波功率,底物配比等因素对丝素蛋白游离氨基酸含量及褐变程度的影响,同时经响应面分析法得出丝素蛋白糖基化最佳反应条件为:底物配比丝素蛋白:β-Cd=2.5:1、微波功率为500w、反应时间为4min,此时优化得到的接枝率为807%。通过实验验证所得接枝率为82.95%,理论值与实际值的相对误差较小。
(2)丝素蛋白糖基化中间产物随加热时间的增加而增加,当加热时间为2min时,中间产物的产生量速率急速上升,当超过4min时中间产物生成量趋于稳定。
(3)探究pH对产物的粒径影响结果表明,在pH为5-6附近时会出现絮凝现象,在该pH范围内不宜应用。
(4)糖基化丝素蛋白相较于纯丝素蛋白颜色偏黄,但不影响后期的吸附应用。
实施例3 SF-Cd的结构表征及微波对其的影响
1.1实验材料
表4-1主要实验材料
Table 4-1 Main experiment materials
2实验方法
2.1产物的溶解度
称取一定量的产物丝素蛋白,加入去离子水溶解,配制成不同pH值得丝素蛋白溶液,在室温下搅拌1h,在10000r/min转速下离心10min,取上层清液,用考马斯亮蓝法测定溶液中的丝素蛋白含量。
2.2荧光光谱分析
将丝素蛋白、β-Cd、微波反应产物在相同条件下分别反应1min、2min、3min、4min,5min、6min、7min其中丝素蛋白和β-Cd为对照组。反应完成后用pH为7.4的磷酸盐缓冲液配成浓度为1mg/mL的溶液待测。在激发波长(λex)为295nm、荧光发射和激发狭缝宽度均为5.0nm的条件下,测定共聚物在305~500nm范围内的荧光发射光谱。Maillard反应所生成的反应产物的特征为激发波长340-370nm,发射波长420-440nm。
2.3巯基和二硫键含量的测定
根据文献,用Ellman试剂法测定巯基
巯基摩尔浓度为:
二硫键计算公式:
其中:A为412nm处的吸光度;ε为消光系数,该系数为13600;D为稀释倍数;C为蛋白质浓度。
2.4红外光谱图
准确称取样品适量,以质量比为1:50的量加入一定量的溴化钾,用研钵研磨至样品和溴化钾充分混合成均匀的粉末,在压片机上将混合粉末压成薄片,再用傅立叶红外分光光度计作全波段(4000-400cm
2.5差热DSC扫描
取3-8mg左右冷冻干燥后的丝素蛋白、环糊精、微波前后丝素蛋白糖基化产物,记下准确质量,放入铝盘中,将样品平铺于铝盘中并用盖子压实,设置温度扫描范围为40-350℃,升温程序:20℃/min。载气N2。载气流量:20mL/min。每次实验前先扫描空铝盘作为空白对照。
2.6扫描电子显微镜分析
取一定量的待测样品,粘在实现剪裁好的导电胶上,用离子溅射镀膜仪在样品表面进行喷金,镀完金膜后将样品放入电子显微镜的放样室中推入观察。
3结果与讨论
3.1巯基和二硫键含量
有研究表明,在丝素蛋白中,小分子蛋白与大分子量蛋白以1:1结合,两者通过二硫键连接而成结构稳定的丝素蛋白,本课题对丝素蛋白中的二硫键及巯基含量进行测定,有助于分析蛋白内部空间延伸状况,结果如图18所示。
分别在水浴(WB)与微波(MW)条件下测定SF+Cd在加热1-7min时的二硫键含量,同时在相同条件下以丝素蛋白作为空白对照实验。可以看出,随着加热时间的延长,二硫键含量均减少。由于水浴加热时间太短,对水浴条件下的产物二硫键含量几乎没有影响;但在微波条件下,即使不加入β-Cd,二硫键含量仍有减少,在加入β-Cd后,产物二硫键含量明显减少。
不同加热方式对糖基化丝素蛋白产物含巯基量的影响如图19所示,可以看出,除水浴加热丝素蛋白组以外,其他三组实验的巯基含量均随加热时间的延长而增加。反应中的巯基含量主要是由二硫键转化而来,当反应超过4min时,SF-Cd(MW)的巯基含量明显下降,主要是由于随着反应的进行,巯基被氧化,可以推测蛋白分子结构并没有发生交联,只是β-折叠结构被打开。水浴加热对巯基含量影响较小,主要是由于加热时间过短。
3.2不同pH和加热方式下的溶解度
由图20我们可以看出,经过微波后的产物溶解性相较微波前有较大差异。SF-Cd(WB)在pH为4的条件下溶解性最低,而SF-Cd(MW)在pH为6左右的条件下溶解性最低,这主要是因为经过糖基化修饰的丝素蛋白的等电点发生了右移,在等电点时,蛋白质分子以两性离子形式存在,由于此时蛋白质分子内部分子净电荷为零,无相同电荷互相排斥,使其极易相互碰撞聚集而产生沉淀。因此在等电点附近的蛋白质溶解性最差。此外,从整体上看,除等电点以外的pH条件下(pH=2-10),经微波糖基化后的丝素蛋白的溶解性能比水浴加热条件下的丝素蛋白要好,说明微波加热条件的效率远高于普通水浴加热。同时通过微波引入糖基,掩蔽了丝素蛋白本身存在的疏水基团,又由于β-Cd表面大量的亲水基团,使得产物的溶解性显著增加。
3.3荧光光谱分析
荧光光谱通过检测蛋白质分子中的内源性氨基酸残基(主要是Trp色氨酸和Tyr酪氨酸)来获取蛋白质的结构变化,从而解释其理化性能的改变。有研究表明,Maillard反应会产出具有荧光特性的物质,该物质的激发波长为340-370nm,发射波长为420-440nm
实验测量了SF和β-Cd分别在水浴7min和微波各时间段的荧光强度,其中测量单丝素蛋白的荧光强度为对照组。研究发现
3.4红外光谱图
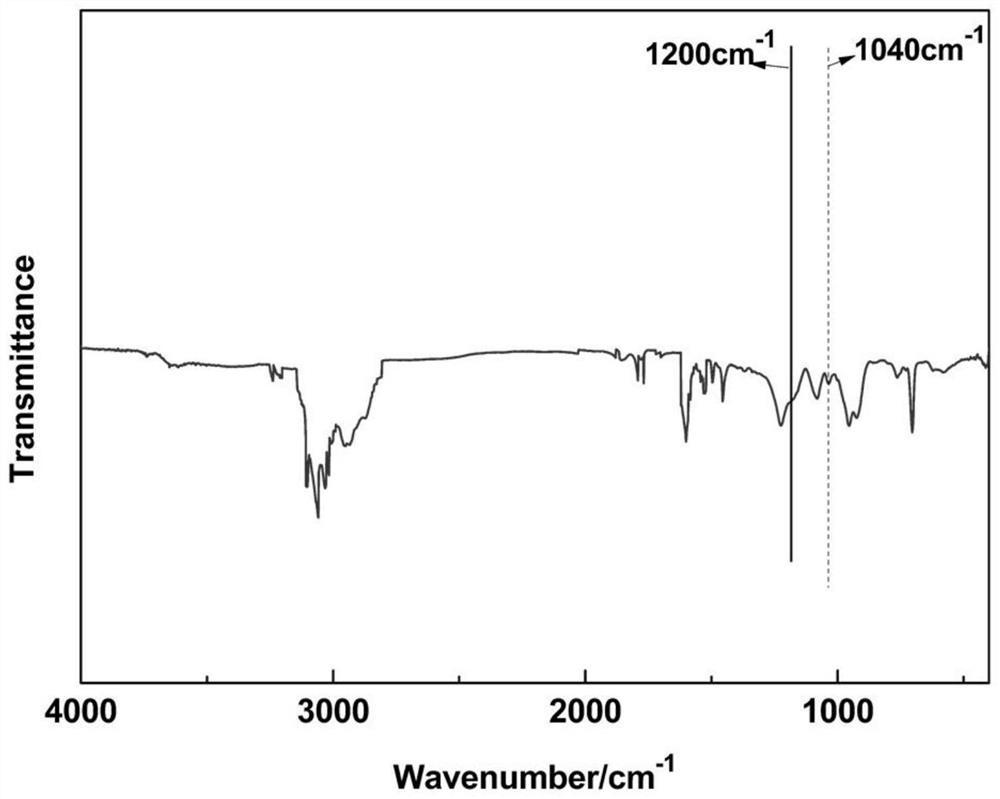
蛋白质和糖类可以通过红外光谱的几个特征吸收峰明显区分开来,特征峰通过不同特征基团的伸缩、弯曲等振动方式体现,故可以利用红外光谱图检验蛋白质和糖类的结合情况。红外结果表明,丝素蛋白的蛋白质特性吸收峰明显,在1650cm
3.5差热DSC扫描
差示扫描量热法(DSC)所测得的温度动态函数可以代表样品的热变性过程。丝素蛋白是一种具有复杂的高级结构的一类高分子物质,热处理会使其从有序的折叠状态向无序的展开状态转变,分子内的作用力减弱,多肽链展开,蛋白构象发生改变,此时由于共价键的重新组合会导致样品吸热
丝素蛋白、环糊精、微波前后产物的DSC曲线如图23所示。
由DSC曲线变化可知,丝素蛋白在182℃处有特征热吸收峰,环糊精在136℃左右有特征热吸收峰,丝素蛋白和环糊精的水浴加热产物在182℃、173℃和135℃左右均出现特征热吸收峰,而丝素蛋白和环糊精微波后的产物在136℃左右无明显的热吸收峰,这说明微波产物中的环糊精与丝素蛋白通过接枝形成Maillard反应产物,只含有一种物质,而水浴产物的反应速率低、反应不彻底导致产物中有三种物质同时存在,图中显示,反应接枝后的复合物的特征热吸收峰在170℃左右处出现,相较丝素蛋白的特征峰向左略有偏移,这可能是因为加入了糖基的丝素蛋白在结构上相较纯丝素蛋白更加无序,折叠结构更加展开。
3.6扫描电子显微镜SEM分析
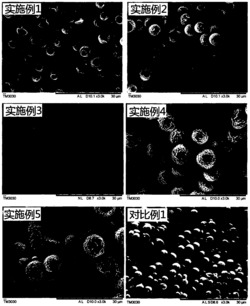
将环糊精、丝素蛋白和糖基化产物经低温冷冻干燥后研碎成粉末状,用扫描电子显微镜观察的外部形态和聚集状态,分别拍摄丝素蛋白、水浴糖基化产物和微波糖基化产物的400倍和2000倍电镜图,结果如图24。观察发现,丝素蛋白粉末颗粒表面光滑平整,且颗颗分明;当经过水浴加热时,产物表面开始粗糙不平,且颗粒变大,颗粒之间发生聚集;当经过微波糖基化加热时,加热产物表面更加粗糙,且微波使丝素蛋白分子聚集成更大的颗粒,这是因为微波的热效应使丝素蛋白的蛋白胶束展开,通过极性分子间的相互摩擦产生热能,这种热效应远强于水浴加热,使丝素蛋白分子间作用剧烈易发生聚集。
4本章小结
(1)经过微波后的产物溶解性相较微波前有较大差异。丝素蛋白和水浴加热后的SF+Cd在pH为4的条件下溶解性最低,而微波后的SF+Cd在pH为6左右的条件下溶解性最低。
(2)荧光光谱结果表明,在激发波长360nm时,丝素蛋白与β-Cd微波糖基化产物发射波长的最强荧光强度在429nm处,微波糖基化后的丝素蛋白荧光强度明显高于水浴加热和未经处理的丝素蛋白,在反应3min时,荧光强度明显增强,表明3-4min时的产物生成速率加快,从而Maillard反应越强烈。
(3)FT-IR图谱表明,丝素蛋白与环糊精通过碳氨缩合反应生成糖基化产物;DSC实验结果说明产物热稳定性良好;SEM图像观察可知丝素蛋白经糖基化后表面粗糙度增加有益于后期的吸附应用。
实施例4体外缓释及机理探究
1实验材料与仪器设备
1.1实验材料
表5-1主要实验材料
Table 5-1 Main experiment materials
2实验方法
2.1L-Ca含量的测定
每个香芹酮(L-Ca)分子上都含有一个羰基,测量L-Ca的含量即测量羰基值。用游离羟铵法测定羰基值,原理如下:中和每克L-Ca与盐酸羟铵反应生成的盐酸所需的KOH的毫克数。盐酸羟铵和KOH的混合物释放的自由羟铵与羰基化合物反应会生成肟
游离羟铵法:
准备以下试剂:
溴酚蓝乙醇溶液:将0.2g溴酚蓝溶于0.1mol/l KOH的乙醇溶液中,加入10mL无水乙醇,煮沸后冷却,用无水乙醇定容至100mL。
盐酸羟铵乙醇溶液:将50g盐酸羟铵溶于100mL水中,加入800mL无水乙醇和10mL溴酚蓝乙醇溶液,用无水乙醇定容至1000mL。观察所形成的分层厚薄,如较薄,加入氢氧化钾乙醇溶液直到溶液变绿;如较厚,加入氢氧化钾乙醇溶液直到溶液变红。检测溶液有效性:加适量盐酸溶液,出现柠檬黄色;加入适量氢氧化钾,出现红色。静置待用。
测量方法:
取3mL含L-Ca的溶液,加入20mL盐酸羟胺乙醇溶液,15mL 0.1mol/L KOH,用盐酸滴定至黄绿色消失。
2.2负载L-Ca的缓释微球的制备
一种微波合成SF-Cd缓释微球的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。














动态评分
0.0