

IPC分类号 : C08G81/00,C08B37/00,C08B37/08,C08H1/00,A61K38/22,A61P3/10,C07K14/575




专利摘要
本发明公开了带有羟胺基的艾塞那肽类似物及其应用,属于医药化学领域。本发明通过对Exendin‑4进行改造,得到具有更长药理作用时间的exendin‑4类似物。与艾塞那肽相比较,本发明的艾塞那肽类似物具有较长药理作用时间的降血糖效果。目标多肽的合成通过正交保护策略固相合成方法实现。
权利要求
1.一种艾塞那肽类似物,其特征在于,是将氨基酸序列如下述(a)所示的多肽中的至少一个赖氨酸的侧链修饰羟胺基-O-NH
(a):His Gly Glu GlyThrPheThr Ser Asp Leu Ser Lys Gln Met Glu GluGlu AlaVal Arg Leu Phe Ile Glu Trp Leu Lys AsnGlyGly Pro Ser SerGly Ala Pro ProProSer-NH
其中,Lys赖氨酸的侧链修饰羟胺基-O-NH
n取自0~10;
所述羟胺基进一步与带有醛基-CHO的化合物相接合;
所述带有醛基-CHO的化合物为多聚糖、带有醛基-CHO的聚乙二醇、带有醛基-CHO的脂肪酸、带有醛基-CHO的聚乙二醇修饰的脂肪酸。
2.根据权利要求1所述的一种艾塞那肽类似物,其特征在于,n=1。
3.根据权利要求1所述的一种艾塞那肽类似物,其特征在于,所述多聚糖包含葡聚糖、多聚唾液酸、糖胺聚糖、聚糖醛酸、透明质酸、麦芽多糖。
4.一种用于治疗糖尿病的药物,其特征在于,含有权利要求1-3任一项所述艾塞那肽类似物。
5.根据权利要求4所述的一种用于治疗糖尿病的药物,其特征在于,所述艾塞那肽类似物与药学上可接受的盐搭配,得到用于治疗糖尿病的药剂。
6.权利要求1-3任一项所述的艾塞那肽类似物在制备治疗或预防糖尿病的药物中的应用。
7.一种改善、预防或治疗肥胖或抑制食欲用药剂学组合物,其特征在于,包含权利要求1-3任一项所述的艾塞那肽类似物作为有效成分。
说明书
技术领域
本发明涉及带有羟胺基的艾塞那肽类似物及其应用,属于医药化学领域。
背景技术
糖尿病是由于机体内胰岛素分泌不足或胰岛素作用缺陷而致使血糖水平增高为特征的代谢异常综合性疾病。持续的慢性高血糖将导致多种组织和器官,如眼、心血管、神经的损伤、功能障碍甚至衰竭。随着全球经济的快速发展,人民生活水平的不断提高,糖尿病已经成为危害人类身体健康的三大非传染性疾病之一。据估计,到2030年,全球糖尿病患者将达到3亿以上。在我国,糖尿病的发病率高达9.7%,2010年的糖尿病患者总数已达到9240万,成为了全球糖尿病患者人数最多的国家。根据糖尿病发病原因的不同,医学上将糖尿病主要分为1型糖尿病和2型糖尿病。
艾塞那肽(Exenatide-4,Ex4)是首个上市用于治疗2型糖尿病的GLP-1类似物药物,Ex4不仅以血糖依赖的方式降低血糖,而且能够促进β细胞增殖。然而该药易被肾脏快速清除,半衰期较短,给患者带来巨大的经济负担和生活不便。因此,开发长效Ex4类似物是当前学术界和药物公司研究热点。
目前采用的提高Ex4半衰期的策略包括对Ex4进行聚乙二醇或者白蛋白修饰。然而,这些药物仍存在着许多缺陷,如造成药物在人体内残留、药效不稳定。
在自然界中,糖基化修饰在增强蛋白质溶解性和稳定性,保持或提高其生物活性,以及延长在体内的半衰期等方面发挥重要作用。另外,糖基化基团具有更好的生物相容性,容易降解且降解产物无毒副作用等优点。但,尚未报道运用糖基化修饰策略来改善Ex4的药物性质。
发明内容
为改善Ex4的药物性质,本发明提供了一类带有羟胺基(羟氨基)-O-NH2键的艾塞那肽(Exendin-4)类似物。
所述带有羟胺基(羟氨基)的艾塞那肽(Exendin-4)类似物是将氨基酸序列如下述(a)、所示的多肽中的至少一个赖氨酸的侧链修饰羟胺基(-O-NH2),
(a):His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu GluGlu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Asn Gly Gly Pro Ser Ser Gly AlaPro Pro Pro Ser-NH2;
其中,Lys赖氨酸的侧链修饰羟胺基(-O-NH2)后的结构为:
n取自0~10,优选1。
在本发明的一种实施方式中,所述带有羟胺基的艾塞那肽类似物上修饰的羟胺基进一步与带有醛基(-CHO)的化合物相接合。所述带有醛基(-CHO)的化合物为多聚糖、带有醛基(-CHO)的聚乙二醇、带有醛基(-CHO)的脂肪酸。所述多聚糖包含葡聚糖、多聚唾液酸、糖胺聚糖、聚糖醛酸、透明质酸、玻尿酸、麦芽多糖。
所述带有羟胺基(羟氨基)-O-NH2键的艾塞那肽(Exendin-4)类似物可与药学上可接受的盐搭配,得到用于治疗糖尿病的药剂。所述药学上可以接受的盐是与盐酸,氢溴酸,硫酸,硝酸或磷酸;琥珀酸,马来酸,醋酸,富马酸,柠檬酸,枸橼酸,酒石酸,苯甲酸,苯磺酸,甲磺酸或萘磺酸形成的盐。所述药剂是任何一种药剂学上所说的片剂、胶囊、酏剂、糖浆、锭剂、吸入剂、喷雾剂、注射剂、膜剂、贴剂、散剂、颗粒剂、块剂、乳剂、栓剂、复方制剂。
本发明在GLP-1受体强效激动剂艾塞那肽的结构基础上,设计合成了赖氨酸类似物,将赖氨酸侧链氨基化学突变成羟胺基团,用于羟胺-醛缀合的进一步长效性修饰,设计合成了一类艾塞那肽类似物。本发明赖氨酸类似物侧链的羟胺基团可以与醛基反应形成肟键,方便高效地引入带有醛基的修饰分子,可避免在早期GLP-1受体长效化激动剂的研发过程中,采用天然赖氨酸作为修饰部位的选择性差、反应不方便等问题。本发明设计了12位、27位赖氨酸两个位点的修饰,既可以单位点修饰,也可以双位点修饰,丰富了艾塞那肽类似物的开发,利于药物的优化和开发。
更重要的是,本发明突破性的采用了羟胺-醛反应形成肟键的缀合方式,使得具有醛基的多糖类化合物可以直接用于修饰,而多糖类化合物一般都具有较好的水溶性,较好的生物兼容性,不同分子量的多糖类化合物都容易获得,如不同分子量范围的葡聚糖、聚唾液酸多糖、透明质酸、玻尿酸、糖胺聚糖、麦芽多糖等。多糖类化合物的修饰可增强缀合物的分子量,很大程度上延长了化合物的半衰期,并可减少化合物的肾脏快速滤过和代谢失活,因此此类化合物的半衰期及体内降糖作用时间显著延长。另外,具有醛基的聚乙二醇化合物、脂肪酸、小分子化合物等,也都可以通过羟胺-醛反应形成肟键的缀合方式与本专利开发的具有羟胺的艾塞那肽类似物进行连接,并发挥延长化合物的半衰期,减少肾脏过滤和代谢失活的作用。
有益效果:
(1)本发明所采用的在艾塞那肽类似物的侧链修饰羟胺基团,易于与醛基反应形成肟键,方便高效地引入带有醛基的修饰分子,用于长效性修饰。
(2)本发明所采用的12、27位赖氨酸突变为带有羟胺基团的赖氨酸衍生物,可以单位点修饰,也可以双位点修饰,丰富了艾塞那肽类似物的开发,利于药物的优化。
(3)本发明所采用的带有羟胺基团侧链的艾塞那肽类似物,易于与带醛基的多糖、聚糖类化合物反应,且直接反应,容易获得,且多糖类化合物具有较好的水溶性、较好的生物兼容性、易获得、分子量选择范围宽等优点。
(4)本发明所获得的艾塞那肽衍生物,经过不同的聚糖或高分子,如葡聚糖、聚唾液酸多糖、透明质酸等,具有与艾塞那肽同等的降糖效果,且半衰期及体内降糖作用时间显著延长。
总之,具有羟胺的艾塞那肽类似物可以用于广泛的药物开发,修饰后的艾塞那肽类似物具有更优的成药性,在更小的给药剂量下,可以达到更优的长效化降血糖效果,能够减少病人多次注射给药的痛苦,提高病人依从性,是2型糖尿病治疗领域中极具开发前景的药物。
附图说明
图1赖类似物K
图2Ex4-12K
图3Ex4-27K
图4Ex4-12,27K
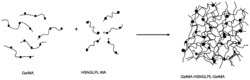
图5葡聚糖修饰Ex4类似物的合成。
图6SDS-PAGE分析(1)Ex4-12K
图7麦芽五糖修饰Ex4类似物。
图8Ex4-12K
图9三种Ex4-dextran复合物的腹腔血糖耐受量试验。LFD:低脂对照组;HFD:高脂对照组;Ex4:艾塞那肽组;Ex4-12(或27,或12,27)K
图10三种Ex4-dextran复合物的血糖抑制长效性实验。
图11.(A)Ex4-12K
图12.三种Ex4-dextran复合物Ex4-12K
图13.三种Ex4-dextran复合物Ex4-12K
具体实施方式
HPLC条件:在Waters 1525型半制备机器上进行纯化,半制备的HPLC使用的是半制备C18(5um,10.0mm×250mm),流动相为乙腈和水(都含有0.1%三氟乙酸),流速3mL·min
实施例1:
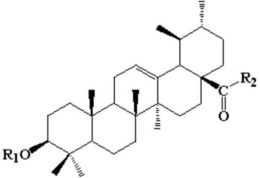
1.1含羟胺侧链的赖氨酸类似物K
如图1所示,Z-Glu-Obzl(1-苄基-N-苄氧羰基-L-谷氨酸)作为合成赖氨酸类似物K
初始原料为商业化的Z-Glu-OBzl,取Z-Glu-Obzl(22.3g,60mmol)溶于干燥四氢呋喃(150mL)。在冰里面加入氯化钠,制成冰盐,通过控制加入氯化钠的量调节冰盐温度,温度调至-10℃左右。将Z-Glu-OBzl溶液放置在-10℃的冰盐浴上搅拌,冷却搅拌10min,注意溶液中防止水进入。加入N-甲基吗啉(N-methylmorpholine,6.6mL,60mmol),在-10℃下搅拌10min,随后再滴加氯甲酸乙酯(ClCO2Et,5.74mL,60mmol),在-10℃下搅拌20min后,再分两次加入硼氢化钠(NaBH4,6.8g,180mmol)。最后在0℃下,用恒压漏斗缓慢滴加甲醇(200mL),滴加1h。滴加完毕后,反应液恢复至室温,并且在室温下搅拌,底物和反应液的TLC通过Hanessuan溶液染色,待反应平衡后,停止搅拌,然后加入1mol·L
取17.6g化合物2溶于150mL二氯甲烷,将该溶液在0℃下搅拌10min,0℃下加入三乙胺(Et3N,2.5eq),随后加入4-甲苯磺酰氯(TsCl,2.5eq)。将该反应液在0℃下搅拌1h,然后使反应液恢复至室温,并在室温下再反应2h。原料和反应液用Hanessuan溶液染色,反应完全后,向反应液中加入300mL二氯甲烷,分别用水、盐水洗涤,收集下层的有机相,用无水硫酸钠干燥。干燥3h后,减压蒸馏去除二氯甲烷,得到粗产物。将得到的粗产物利用硅胶柱(乙酸乙酯∶石油醚=1:8)进行纯化,得到油状的产物即化合物3。
将N-羟基邻苯二甲酰亚胺(NOP,2.0eq)充分溶解于20mL N,N-二甲基甲酰胺,再取1,8-二氮双环[5.4.0]十一-7-烯(DBU,2eq)溶解于10mL N,N-二甲基甲酰胺。在0℃下,将1,8-二氮双环[5.4.0]十一-7-烯溶液加入到N-羟基邻苯二甲酰亚胺溶液中,该混合液在0℃下搅拌30min,然后将化合物3(1.0eq)溶解于N,N-二甲基甲酰胺溶液,在0℃下,将化合物3的N,N-二甲基甲酰胺溶液滴加在上述的N-羟基邻苯二甲酰亚胺和1,8-二氮双环[5.4.0]十一-7-烯混合液中。反应液恢复至室温,再搅拌反应。原料和反应液用Hanessuan溶液染色,待反应完全后,向反应液中加入300mL乙酸乙酯,然后用水和盐水各清洗萃取液两次,以洗掉反应液中的N,N-二甲基甲酰胺,用无水硫酸钠干燥萃取液。干燥3h后,减压蒸馏抽除乙酸乙酯,得到粗产物。将粗产物利用乙酸乙酯和石油醚进行重结晶,得到白色固体,即化合物4。
称取18.1g化合物4用20mL甲醇充分溶解,在0℃下搅拌10min,加入水合肼(10eq,用甲醇配置成0.2mol·L
将化合物5用30mL二氯甲烷充分溶解,向该溶液中加入三乙胺(1.2eq),将该混合液在0℃下冷却搅拌20min。取Boc酸酐(Boc2O,1.5eq)溶于50mL二氯甲烷中,然后在0℃下,将Boc酸酐溶液滴加在上述混合液中,滴加完成后,将反应液恢复至室温,并在室温下搅拌过夜。原料和反应液用5%茚三酮染色,待反应完全,向反应液中加入150mL二氯甲烷,用二氯甲烷萃取反应液,然后用水和盐水洗涤萃取液,用入无水硫酸钠干燥萃取液,干燥3h后,减压蒸馏抽去二氯甲烷,得到粗产物,将粗产物利用硅胶柱(乙酸乙酯∶石油醚=1:8)进行纯化,得到油状产物,即化合物6。
取10.8g化合物6,溶于10mL甲醇中,加入1.1g氢氧化钯/碳,将反应瓶抽真空,在瓶子上插一个氢气球,在室温下搅拌过夜。待反应完全,利用硅藻土将反应液中的氢氧化钯/碳过滤去除,并用甲醇洗涤滤渣,收集滤液,减压蒸馏抽掉甲醇,即可以将化合物6上面的氨基保护基Cbz基团和羧基保护基Bn基团脱去,得到的产物无需进一步纯化,可以直接用于下一步反应。取该化合物溶于100mL 1,4-二氧六环和水(1:1,v/v)中,在冰浴条件下,加入1.5eq的碳酸氢钠粉末,在冰浴下再搅拌10min。取1.5eq的芴甲氧羰酰琥珀酰亚胺溶于1,4-二氧六环,在0℃下,用恒压漏斗将芴甲氧羰酰琥珀酰亚胺溶液逐渐滴加入到上述的溶液中去,滴加完成后,恢复至室温再反应4h。待反应完全后,减压蒸馏抽去1,4-二氧六环,加入盐酸,调节pH至2,向反应液中加入100mL的乙酸乙酯,用乙酸乙酯萃取三次,收集萃取液,用5%柠檬酸洗涤两次,再用水和盐水各洗涤一次。收集乙酸乙酯溶液,用无水硫酸钠干燥。干燥3h后,减压蒸馏抽去乙酸乙酯,得到粗产物,最后经过硅胶柱(乙酸乙酯∶石油醚=1:6)分离,得到纯产物,真空干燥为晶体状固体即最终的目标化合物7(赖氨酸类似物K
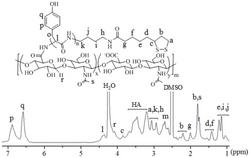
1.2含K
基于Fmoc保护策略化学,从Ex4的C端到N端将氨基酸依次连接在树脂上。使用的树脂为Rink MBHA Resin(负载量:0.217mmol·g
Ex4-12K
Ex4-27K
Ex4-12,27K
利用上述的“一步缓慢梯度法”纯化三种Ex4类似物,纯化后的产物通过HPLC和MALDI-TOF MS分析鉴定。得到Ex4-12K
得到Ex4-27K
得到Ex4-12,27K
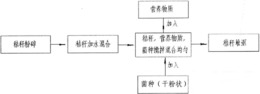
1.3.葡聚糖修饰的艾塞那肽衍生物的合成和活性
利用Ex4类似物(Ex4-12K
具体地,反应的缓冲液:100mmol·L
如图6所示:Ex4-12K
通过实验证明,本发明成功合成了葡聚糖修饰的艾塞那肽化合物Ex4-12K
1.4麦芽五糖糖基化修饰Ex4类似物
麦芽五糖对Ex4类似物进行糖基化修饰过程如图8所示,修饰方法同1.3、图5,将麦芽五糖分别对三种Ex4类似物进行糖基化修饰,验证这三种Ex4类似物进行糖基化修饰的可行性。
具体地,反应的缓冲液:100mmol·L-
Ex4-12K
1.5 Ex4-dextran复合物的活性
小鼠的血糖耐受量试验结果如图9所示:
在給小鼠注射葡萄糖25min后,各组小鼠血糖浓度达到了高峰,随后所有组分的小鼠的血糖都开始下降。除了高脂对照组小鼠血糖浓度一直维持在很高的水平外,结果表明,葡聚糖修饰的三种Ex4类似物Ex4(12K
1.6 Ex4-dextran-20000的合成与活性
我们使用分子量为20000的葡聚糖对上述类似物(Ex4-12K
具体地,反应的缓冲液:100mmol·L
在25℃下反应24h后,通过SDS-PAGE监测反应。SDS-PAGE中含有0.1%SDS,凝胶的浓度为12%,在120v的恒压电流下跑2h。然后浸入考马斯亮蓝R250进行染色,染色完成后,脱色三次,每次脱色40min。
我们利用SDS-PAGE监控葡聚糖修饰Ex4类似物的反应,并对最终糖肽复合物纯品进行凝胶色谱表征。具体实验结果如下:
经过HPLC分离后,得到是Ex4-12K
以同样的方法制得:Ex4-27K
经过HPLC分离,最终通过Sephadex G-25分离,除去剩余未反应Ex4-12K
在給小鼠注射葡萄糖25min后,各组小鼠血糖浓度达到了高峰,随后所有组分的小鼠的血糖都开始下降。除了高脂对照组小鼠血糖浓度一直维持在很高的水平外,结果表明,葡聚糖修饰的三种Ex4类似物Ex4-12K
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
SEQUENCE LISTING
<110> 江南大学
<120> 带有羟胺基的艾塞那肽类似物及其应用
<160> 1
<170> PatentIn version 3.3
<210> 1
<211> 39
<212> PRT
<213> 人工序列
<400> 1
His Gly Glu Gly Thr Phe Thr Ser Asp Leu Ser Lys Gln Met Glu Glu
1 5 1015
Glu Ala Val Arg Leu Phe Ile Glu Trp Leu Lys Asn Gly Gly Pro Ser
202530
Ser Gly Ala Pro Pro Pro Ser
35
带有羟胺基的艾塞那肽类似物及其应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。










动态评分
0.0