

IPC分类号 : C07J41/00I,C12P33/20I,A61K9/127I,A61K47/28I,A61K31/337I,A61K31/4525I,A61P35/00I




专利摘要
本发明涉及一种肝癌靶向糖配体分子及其制备方法和载药系统,特别是涉及肝脏ASGPR受体靶点的靶向糖配体分子,以及应用该靶向糖配体分子制备的脂质体。该糖配体分子由三部分构成:肝靶向糖基团、亲脂性“锚”和通过酯键连接两者的取代或非取代的以‑(CH2)8直链间隔臂。该肝癌靶向糖配体分子能够使得该载药系统中的抗肿瘤药物最大限度浓集于肝肿瘤细胞,提高药物治疗指数,降低全身毒副作用,改善患者的生存质量。
权利要求
1.一种肝靶向糖配体分子,由三部分构成:肝靶向糖基团、亲脂性“锚”结构和通过酯键连接两者的非取代的以-(CH2)
2.如权利要求1所述的肝靶向糖配体分子,其特征在于,所述亲脂性“锚”结构镶嵌于载药体统中,通过所述-(CH
3.一种权利要求1-2任一项所述的肝靶向糖配体分子的制备方法,其特征在于,在非水相中通过生物酶催化法构建糖配体分子。
4. 如权利要求3所述的肝靶向糖配体分子的制备方法,其以胆固醇-癸二酸单烯酯(CHS-SE)为起始原料,在脂肪酶TL IM催化下,与GalNAc上的C
说明书
本申请是中国专利申请201611167938.8的分案申请,原申请的申请日为2016 年12月16日,发明名称为肝癌靶向糖配体分子及其制备方法和载药系统。
技术领域
本发明涉及一种肝癌靶向糖配体分子及其脂质体的制备,特别是涉及肝脏 ASGPR受体靶点的靶向糖配体分子,以及应用该靶向糖配体分子制备的脂质体。
背景技术
原发性肝癌(primary liver cancer,PLC)是临床上最常见的消化道恶性肿瘤 之一,其中90%为肝细胞肝癌(hepatocellular carcinoma,HCC)。我国为HCC高 发国,约占世界HCC发生总数的70%,占国内肿瘤死亡率第3位[Chen,W.,et al., Cancer statisticsin China,2015.CA Cancer J Clin,2016.66(2):p.115-32]。早期 HCC首选治疗方法包括手术切除和肝移植在内的外科治疗。但大多数患者确诊 时已是中晚期,且常合并乙/丙型病毒性肝炎、肝硬化等,已失去外科手术机会, 而此时化疗常常是唯一有效的治疗手段。然而化疗药物普遍存在疗效低、副作用 多等诸多缺点。除化疗药物本身药理作用尚不够理想外,其主要原因就是不能将 药物准确有效地转运至肝脏病灶部位,导致药物生物利用度低下;即使药物能够 靶向运输到肝脏,又被肝脏迅速代谢,加大剂量和缩短用药时间间隔又导致对其 他正常器官严重毒副作用,大大降低了药物的有效作用。因以提高癌变部位的药物浓度、延长药物作用时间和降低药物毒副作用为目的的新型纳米靶向控释系统 在肝癌药物治疗研究中的应用,逐渐成为肝癌治疗研究的热点。
肝实质细胞表面存在大量的受体,例如去唾液酸糖蛋白受体(asialoglycoprotein receptor,ASGPR)、转铁蛋白受体(transferrin receptor,TfR)、高密度脂蛋白受体(high density lipoprotein receptor,HDLR)、低密度脂蛋白受体(low density lipoprotein receptor,LDLR)、生长因子受体、胰岛素受体等,其中 ASGPR和TfR是两种高效的内吞受体,TfR存在于许多种细胞的细胞膜上, 而ASGPR只存在于肝实质细胞,因此ASGPR成为肝脏定向转运的最佳受体[黄 渊余、梁子才,去唾液酸糖蛋白受体及其在药物肝靶向递送中的应用.生物化学与 生物物理进展,2015(06):第501-510页]。
ASGPR是数量丰富的一种异源低聚物的内吞型受体,只存在于肝脏实质细 胞朝向窦状隙一侧的细胞膜表面,能特异性地识别与结合具有D-半乳糖 (D-galactose,Gal)或N-乙酰半乳糖胺(N-acetylgalactosamine,GalNAc)作为末端 糖基的配体分子[Franssen,E.J.F.,et al.,Hepatic and intrahepatic targeting of an anti-inflammatoryagent with human serum albumin and neoglycoproteins as carriermolecules.Biochemical Pharmacology,1993.45(6):p.1215-26]。文献研究发现, ASGPR受体与配体的亲和性主要受以下因素影响:1)配体末端糖分子类型。末 端为Gal或GalNAc的配体分子均可以被ASGPR识别,GalNAc与ASGPR结 合的亲和性比Gal高10~50倍[Rensen,P.C.,et al.,Determination of the upper size limit for uptake and processingof ligands by the asialoglycoprotein receptor on hepatocytes in vitro and invivo.J Biol Chem,2001.276(40):p.37577-84],而且 GalNAc配体分子更容易逃逸Kupffer细胞的识别而更多靶向于肝肿瘤细胞 [D'Souza,A.A.and P.V.Devarajan,Asialoglycoprotein receptor mediated hepatocyte targeting-strategies andapplications.J Control Release,2015.203:p.126-39];2) 糖分子取代位点与ASGPR受体亲和性有如下规律:Gal(或GalNAc)上的1-OH、 2-OH(或2-amino)、3-OH、4-OH和5-CH2-都参与ASGPR受体的结合;5-CH2- 连有负电性基团或4-OH、3-OH、2-OH(或2-amino)被取代后,与受体亲和力 急剧下降;1-OH发生苷化取代时,当糖苷键为α构象时,与受体亲和力减弱,β构象时且取代基为直链结构时,亲和力不受影响[Lee,R.T.,Binding site of the rabbitliver lectin specific for galactose/N-acetylgalactosamine.Biochemistry,1982.21(5):p.1045-50];只有6-OH不参与受体结合且指向溶剂区,因而适合作为连 接位点与载体、药物等相连[Stokmaier,D.,et al.,Design,synthesis and evaluation ofmonovalent ligands for the asialoglycoprotein receptor(ASGP-R).Bioorg MedChem, 2009.17(20):p.7254-64];然而,使用不同的末端分子,间隔臂的长度与亲和力 之间的关系并没有更详细的数据披露。
因此,根据上述HCC治疗现状,本发明提供一种与肝细胞ASGPR受体特 异性结合的靶向糖配体分子,同时,该配体分子能够修饰特定载药系统,借助该 载药系统使抗肿瘤药物最大限度浓集于肝肿瘤细胞,减少药物在其他正常组织分 布,显著提高药物治疗指数,降低全身毒副作用,改善患者的生存质量。
另外,根据文献报道,糖基配体分子因为存在大量的糖羟基,采用化学法合 成需要非反应位点的羟基进行精确的保护和脱保护措施,使得反应条件复杂,步 骤冗长,效率低下。而生物酶是一种具有催化功能的蛋白质,与化学催化剂相比, 具有高效、高区域选择性、低毒等优点,被称之为绿色催化剂,特别是在糖分子 的区域选择性酯化上具有独特的优势。因此,本发明提供一种高效专一性强的生 物酶合成糖基配体分子的方法。
发明内容
本发明提供一种肝靶向糖配体分子,该糖配体分子特定加载于纳米载药系 统,能够使得该载药系统中的抗肿瘤药物最大限度浓集于肝肿瘤细胞,著提高药 物治疗指数,降低全身毒副作用,改善患者的生存质量。该肝靶向糖配体分子由 三部分构成:肝靶向糖基团、亲脂性“锚”结构和通过酯键连接两者的取代或非取 代的以-(CH2)8直链间隔臂。
进一步,本申请所述肝靶向基团,包括但不限于GalNAc、亲脂性“锚”包括 但不限于胆固醇(cholesterol,Chol)及其结构类似物;
进一步,申请人惊讶发现,当间隔臂为直链非取代的-(CH2)8更适合加载于 载药系统。
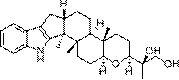
具体来说,本发明提供一种乙酰氨基半乳糖(N-acetylgalactosamine,GalNAc)配体分子,该配体分子能够特异性与肝细胞中ASGPR结合。该配体分子结构如 下:
本发明还提供一种制备该糖配体分子的方法,即在非水相中通过生物酶催化 法构建糖配体分子,具体来说,是构建GalNAc配体分子。
具体实验方法为,以胆固醇-癸二酸单烯酯(CHS-SE)为起始原料,在脂肪 酶TL IM催化下,高选择性专一与GalNAc上C6-OH酯化偶联,即得到肝靶向 配体材料GalNAc-C8-Chol。
具体试验步骤如下:取10mL具塞小瓶,加入CHS-SE(0.1mmol)、GalNAc(0.05mmol),再加入5mL脱水丙酮,加酶TL IM 20mg,空气浴恒温振荡器内 振荡(45℃,250r·min
发明人经过大量实验发现,脂肪酶TL IM在非水相溶剂中可专一催化 GalNAc上C6-OH酯化反应,且反应条件温和,转化率高,纯化步骤简便,所用 反应试剂低毒易去除,合成材料廉价易得。
本发明还提供一种肝靶向配体分子的载药系统,所述肝靶向配体分子的亲脂 性“锚”结构镶嵌于所述载药体统中,通过取代或非取代的以-(CH2)8直链间隔 臂将肝靶向糖基团暴露于载药体统表面。
进一步,所述载药系统包括但不限于纳米脂质体载药系统。载药系统为纳米 脂质体载药系统时,所述亲脂性“锚”结构镶嵌于所述纳米脂质体载药系统的磷 脂双分子层中。
具体来说,是GalNAc配体分子修饰的载药系统,则该GalNAc配体分子暴 露于载药体统表面。
发明人惊讶地发现,当间隔臂为直链非取代的-(CH2)8更适合的糖配体分子, 具体来说,是GalNAc配体分子时,所修饰的脂质体载药系统更稳定,与ASGPR 亲和力最高。所述的GalNAc配体修饰的纳米脂质体载药系统如图9所示。
本发明还提供一种生物酶催化法,在非水相中构建GalNAc修饰的纳米脂质 体肝肿瘤靶向递药系统。
具体制备方法为:按一定比例称取磷脂(PC)、胆固醇(CHS)、DSPG-Na、 GalNAc配体分子、药物等混合溶解于氯仿中,在茄形瓶中55℃、40rpm旋蒸除 去氯仿,形成均匀脂膜,加入pH 7.4PBS缓冲液,55℃水化1h,然后依次用100 nm、50nm滤膜挤压滤过,即得均一纳米粒。
实验结果表明,该GalNAc修饰的脂质体摄取率更高。
进一步,所述纳米脂质体肝肿瘤靶向递药系统所包载药物为抗癌化学小分子 药物和/或中药单体药物和/或生物利用度增强剂,包括但不限于药物紫杉醇 (paclitaxel,PTX)和/或生物利用度增强剂胡椒碱。
附图说明
图1 GalNAc-C8-Chol质谱图
图2 GalNAc-C8-Chol13C NMR谱图
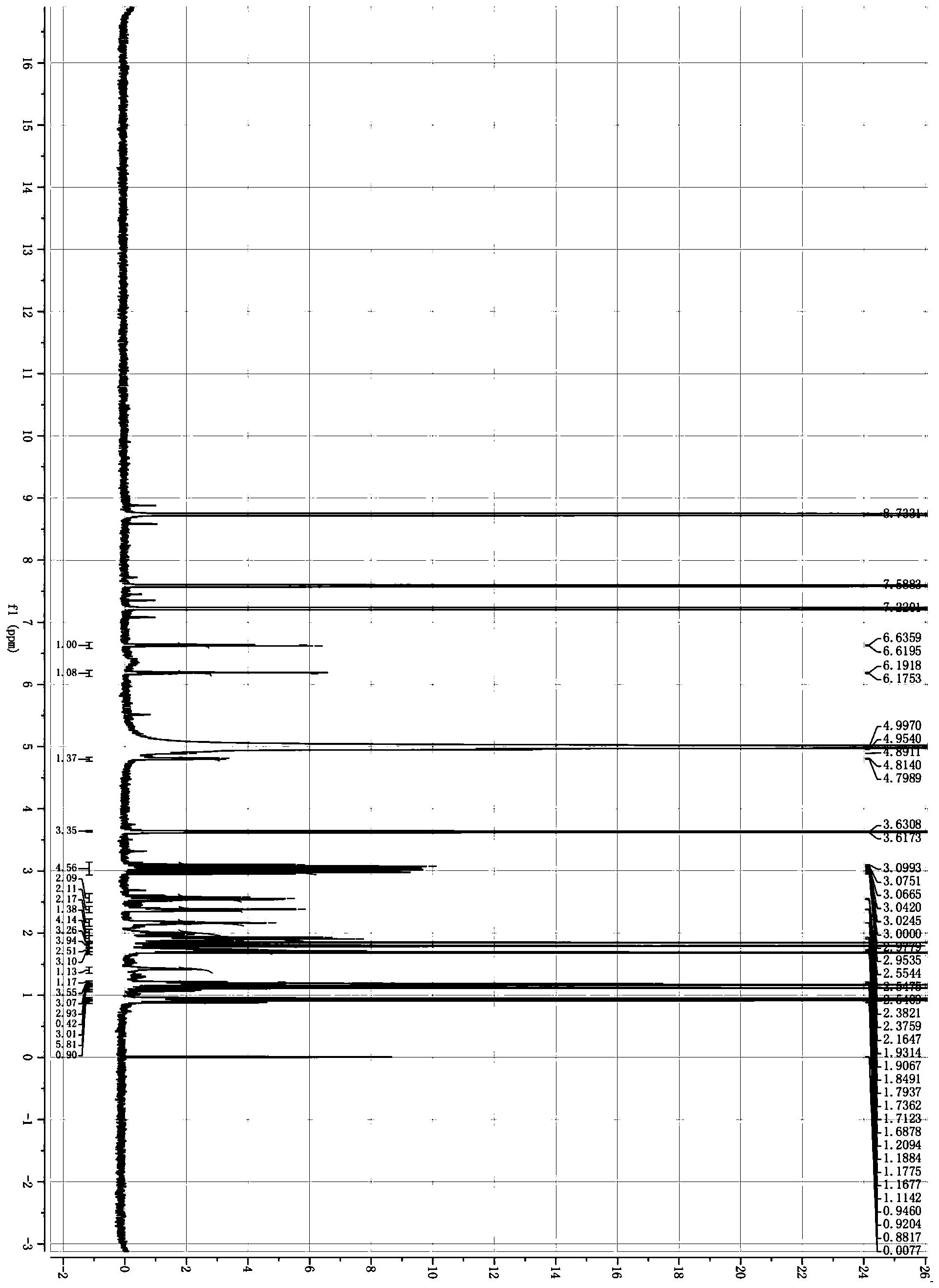
图3 GalNAc-C8-Chol1H NMR谱图
图4 GalNAc(A)与GalNAc-C8-Chol糖基部分(B)13C-NMR谱图比较
图5 GalNAc-C8-Chol部分HMBC谱图
图6 GalNAc修饰脂质体(A)和普通纳米脂质体(B)粒径分布
图7 GalNAc修饰脂质体(A)和普通纳米脂质体(B)Zeta电位分布
图8 HepG2细胞对FL-LP、GAL-FL、GalNAc-LP-FL摄取率比较(n=3)。(**p< 0.01)。
图9 GalNAc配体修饰的纳米脂质体载药系统。
下面通过具体实施方式对本发明作进一步说明。
具体实施方式
下面列举一部分具体实施例对本发明进行说明,有必要在此指出的是以下 具体实施例只用于对本发明作进一步说明,不代表对本发明保护范围的限制。 其他人根据本发明做出的一些非本质的修改和调整仍属于本发明的保护范围。
实施例1 GalNAc-C8-Chol的合成
方法:取10mL具塞小瓶,加入CHS-SE(0.1mmol)、GalNAc(0.05mmol), 再加入5mL脱水丙酮,加酶TL IM 20mg,空气浴恒温振荡器内振荡(45℃,250 r·min-1),反应24h。待反应结束后,过滤除酶,回收溶剂,即得产物,反应方 程式见图1所示。产物通过快速硅胶柱层析纯化,纯化产物利用红外、ESI、1H NMR、13C NMR进行结构鉴定。
合成产物鉴定
产物结构经MS、NMR鉴定为目标产物,具体数据如下:
MS条件:产物用适量甲醇溶解后,用质谱分析仪(MS)分析,质谱参数: 三重四级杆LC-MS/MS:电喷雾正离子化(ESI+)检测,扫描范围为m/z150-m/z 1000,[M+Na]+:796.50;
1H NMR(500MHz,C5D5N)δ:5.99(H-1'α,d,J=3.65,3.65Hz,1H),5.43 (H-2,d,J=5.19Hz,1H),5.33(H-1'β,m,1H),4.93(H-3,t,J=4.91,4.91Hz, 1H),4.91(H-6'α,d,J=2.13Hz,1H),4.90(H-2',s,1H),4.89(H-5',s,1H), 4.86(H-6'β,m,1H),4.64(H-3',d,J=10.74Hz,1H),4.50(H-4',s,1H),2.54 (H-7,m,2H),2.41(H-28,t,J=7.43,7.43Hz,2H),2.33(H-35,td,J=1.59, 7.36,7.25Hz,2H),2.13(H-38,s,3H),2.03-1.07(m,38H),1.03(H-24,s, 3H),0.99(H-25,d,J=6.52Hz,3H),0.91(H-22*2,dd,J=1.14,6.61,6H), 0.68(H-26,s,3H);13C NMR(125MHz,C5D5N):δ:173.94(C-36),173.46(C-27), 171.34(C-37),140.43(C-1),123.25(C-2),93.30(C-1'),74.27(C-3),70.84(C-4'), 69.93(C-5'),69.56(C-3'),65.68(C-6'),57.17(C-4),56.74(C-5),52.59(C-2'), 50.64(C-6),42.88(C-8),40.32(C-9),40.13(C-10),39.02(C-7),37.64(C-11), 37.23(C-12),36.88(C-13),36.44(C-14),35.15(C-28),34.73(C-35),32.55(C-15), 32.42(C-16),29.72(C-31,32),29.68(C-30,33),28.91(C-18),28.65(C-19),28.61(C-17),25.75(C-29),25.56(C-34),24.89(C-20),24.55(C-21),23.67(C-38), 23.35(C-22),23.09(C-22),21.67(C-23),19.78(C-24),19.35(C-25),12.39(C-26)。
质谱图、核磁图见图1、2、3
为验证CHS-SE与GalNAc在TL IM脂肪酶催化下酯化的位点,我们对比了
为进一步确证CHS-SE与GalNAc在TL IM催化下酯化的位点,我们运用 NMR波谱技术(HMBC)对结构中糖C-6位氢与羰基(C-36)进行异核远程相 关性分析,发现C-6位的两个氢(H-6'a,H-6'b)与C-36位羰基碳有远程相关(见 图5),进一步证实了酯化反应只存在于糖基C-6位羟基。
实施例2 GalNAc修饰纳米脂质体的制备和表征
制备方法:按一定比例称取磷脂(PC)、胆固醇(CHS)、DSPG-Na、GalNAc配 体分子、药物等按(40:20:1:4:1)质量比混合溶解于氯仿中,在茄形瓶中 55℃、40rpm旋蒸除去氯仿,形成均匀脂膜,加入pH 7.4PBS缓冲液,55℃水 化1h,然后依次用100nm、50nm滤膜挤压滤过,即得均一纳米粒。
并对两者的粒径(图6)、zeta电位(图7)、包封率等进行表征。结果使用该 法制得纳米粒径较小,粒径分布均匀,质量稳定,包封率高(见表1),满足后继 试验要求。
表1普通脂质体与半乳糖修饰纳米脂质体的比较(n=3)
实施例3 GalNAc修饰纳米脂质体体外靶向性研究
实验方法:取处于对数生长期的人肝癌HepG2细胞接种于24孔板内,待细 胞融合度达80%左右且细胞形态饱满后,分别加入适量普通荧光脂质体(FL-LP) 和GalNAc-LP-FL(用培养基调整脂质浓度为1mmol·mL
通过人肝癌HepG2细胞评价GalNAc修饰荧光脂质体(GalNAc-LP-FL)肝 靶向性,结果显示HepG2对经GalNAc修饰后的脂质体摄取率显著高于普通脂 质体(FL-LP)。预先加入的GalNAc可显著抑制HepG2对GalNAc修饰脂质体 的摄取。而且我们还对比了前期采用半乳糖(GAL)修饰脂质体(GAL-FL)与 GalNAc修饰脂质体的摄取率差异,发现HepG2细胞对GalNAc修饰脂质体摄取 率更高,见图8。
肝癌靶向糖配体分子及其制备方法和载药系统专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。













动态评分
0.0