










专利摘要
具有抗肿瘤活性的砷糖化合物及其制法和应用。本发明涉及N-(4”-(1”,3”,2”-二硫砷烷)苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺。由阿散酸经过二氧化硫还原,盐酸成盐,碱化,上保护基制备4-(1,3,2-二硫砷烷)苯胺,然后与炔丙酸在N,?N’-环己基碳二亚胺作用下生成N-(4-(1,3,2-二硫砷烷)苯基)炔酰胺;2-叠氮-1,3,4,6-O-乙酰基-D-葡萄糖与N-(4-(1,3,2-二硫砷烷)苯基)炔酰胺反应生成N-(4”-氧化砷苯基)-1-(1’,3’,4’,?6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺;所得产物再与1,2-乙二硫醇反应得到本发明所述化合物。
权利要求
1.N-(4”-(1”,3”,2”-二硫砷烷)苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺I,其结构式如下:
。
2.权利要求1所述化合物I的合成方法,其特征是包括以下步骤:
步骤一:以阿散酸为原料,在二氧化硫与碘化钾的作用下还原成三价砷化物后,经过盐酸酸化成4-二氯砷苯胺盐酸盐;4-二氯砷苯胺盐酸盐经过以下两种方法之一制备4-(1,3,2-二硫砷烷)苯胺;方法一:在碱性条件下生成对氨基苯基氧化砷,再进一步在溶剂中与1,2-乙二硫醇反应生成4-(1,3,2-二硫砷烷)苯胺;方法二:4-二氯砷苯胺盐酸盐直接在碱性条件下与1,2-乙二硫醇反应生成4-(1,3,2-二硫砷烷)苯胺;
步骤二:以4-(1,3,2-二硫砷烷)苯胺为原料,与炔丙酸在N,N’-环己基碳二亚胺作用下生成N-(4-(1,3,2-二硫砷烷)苯基)炔酰胺;
步骤三:2-叠氮-1,3,4,6-O-乙酰基-D-葡萄糖中间体与N-(4-(1,3,2-二硫砷烷)苯基)炔酰胺在溶剂中在一价铜催化下发生点击反应生成N-(4”-氧化砷苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺;
步骤四:N-(4”-氧化砷苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺在溶剂中与1,2-乙二硫醇反应生成产物N-(4”-(1”,3”,2”-二硫砷烷)苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺I。
3.根据权利要求2所述的合成方法,其特征是:步骤二中4-(1,3,2-二硫砷烷)苯胺与丙炔酸在N,N’-环己基碳二亚胺作用下生成N-(4-(1,3,2-二硫砷烷)苯基)炔酰胺;所用的溶剂是二氯甲烷、N,N-二甲基甲酰胺或四氢呋喃。
4.根据权利要求2所述的合成方法,其特征是:步骤三的反应条件是在氮气保护的条件下,在体积比为1:1的叔丁醇与水混合溶剂或者N,N-二甲基甲酰胺中100℃反应4小时,或者在微波合成仪中反应30min。
5.根据权利要求2所述的合成方法,其特征是:步骤四的反应条件是在甲醇中室温搅拌4小时。
6.根据权利要求2所述的合成方法,其特征是:步骤一中经过盐酸酸化成4-二氯砷苯胺盐酸盐后,经过过滤和干燥后再用于制备4-(1,3,2-二硫砷烷)苯胺;步骤一方法一中,4-二氯砷苯胺盐酸盐先在碱性条件下水解后,经过过滤、干燥,再在甲醇中与1,2-乙二硫醇反应,经过重结晶得到4-(1,3,2-二硫砷烷)苯胺;步骤一方法二中,4-二氯砷苯胺盐酸盐在碱性条件下直接与1,2-乙二硫醇反应,经过重结晶得到4-(1,3,2-二硫砷烷)苯胺。
7.根据权利要求2所述的合成方法,其特征是:步骤二反应完成后,经过柱层析得到N-(4-(1,3,2-二硫砷烷)苯基)炔酰胺。
8.根据权利要求2所述的合成方法,其特征是:步骤三的后处理经过硅藻土过滤,减压蒸馏,柱层析得到N-(4”-氧化砷苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺。
9.根据权利要求2所述的合成方法,其特征是:步骤四反应完成后将反应体系浓缩,析出晶体或者柱层析,干燥得到N-(4”-(1”,3”,2”-二硫砷烷)苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺I。
10.权利要求1所述化合物在制备抗直肠癌的药物中的应用。
说明书
技术领域
本发明涉及具有抗肿瘤活性的化合物N-(4”-(1”,3”,2”-二硫砷烷)苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺及其制备方法及其应用。
本发明提供具有良好抗直肠癌活性的化合物N-(4”-(1”,3”,2”-二硫砷烷)苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺及其制备方法及其应用。
背景技术
直肠癌是指从齿状线至直肠乙状结肠交界处之间的癌,是消化道最常见的恶性肿瘤之一。直肠癌位置低,容易被直肠指诊及乙状结肠镜诊断。目前,直肠癌的治疗需要以外科手术为主,辅以化疗、放疗的综合治疗。对于手术治疗,因其位置深入盆腔,解剖关系复杂,手术不易彻底,术后复发率高。中下段直肠癌与肛管括约肌接近,手术时很难保留肛门及其功能是手术的一个难题,也是手术方法上争论最多的一种疾病。化疗、放疗在杀死癌细胞的同时也会将正常细胞杀死,还能导致胃肠功能紊乱、骨髓抑制等副作用,大大降低了患者的生存质量。因此,寻找一种代替或部分代替手术、化疗、放疗方法的药物,具有十分重要的意义。
本发明涉及的化合物N-(4”-(1”,3”,2”-二硫砷烷)苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺I经过检测具有良好的抗肿瘤活性,是未见文献报道的新化合物。本发明所用的原料为2-氨基葡萄糖是可以来自壳聚糖降解的产物。壳聚糖在动物体内的降解成为2-氨基葡萄糖。壳聚糖也有文献报道具有抗肿瘤的生物活性。文献报道1,4位取代的1,2,3-三唑具有良好的抗肿瘤活性。在三唑衍生物的4位引入对氨基苯基氧化砷(3价)及其相应的衍生物。文献报道三价的砷化物具有良好的抗肿瘤活性,但是细胞毒性较大。最近文献报道壳聚糖可以减低三价砷化物的毒性,同时能够协同增强抗肿瘤活性。2-氨基葡萄糖是壳聚糖的单体,对2-氨基进行叠氮化的修饰后,可通过点击反应引入三价砷化物。1,2-乙二硫醇保护的三价砷化物就有靶向定位,同时又可以保护三价砷化物。文献报道天然砷糖是无毒的。
发明内容
本发明的目的是提供一种化合物及其制备方法和用途,该化合物具有良好的抗肿瘤效果。
本发明提供的技术方案是:N-(4”-(1”,3”,2”-二硫砷烷)苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺(化合物I),其结构式为:
。
本发明还提供了上述化合物I的合成方法,包括以下步骤:
步骤一:以阿散酸为原料,经过二氧化硫还原,盐酸成盐,碱化,上保护基制备4-(1,3,2-二硫砷烷)苯胺;或者以阿散酸为原料,经过二氧化硫还原,盐酸成盐,直接在碱性条件下上保护基团制备4-(1,3,2-二硫砷烷)苯胺。
步骤二:以4-(1,3,2-二硫砷烷)苯胺为原料,与炔丙酸在N,N’-环己基碳二亚胺作用下生成N-(4-(1,3,2-二硫砷烷)苯基)炔酰胺。
步骤三:2-叠氮-1,3,4,6-O-乙酰基-D-葡萄糖中间体与N-(4-(1,3,2-二硫砷烷)苯基)炔酰胺在溶剂中在一价铜催化下发生点击反应生成N-(4”-氧化砷苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺。
步骤四:N-(4”-氧化砷苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺在溶剂中与1,2-乙二硫醇反应生成产物N-(4”-(1”,3”,2”-二硫砷烷)苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺I。
上述步骤一中制备4-二氯砷苯胺盐酸盐所用的溶剂是甲醇、四氢呋喃之一。反应温度是室温。制备4-(1,3,2-二硫砷烷)苯胺所用的溶剂是甲醇或碳酸钠水溶液。其中运用4-二氯砷苯胺盐酸盐通过氨水碱化,析出晶体后过滤,经过干燥之后,溶于甲醇或四氢呋喃之中,与1.0当量的1,2-乙二硫醇反应生成4-(1,3,2-二硫砷烷)苯胺;运用4-二氯砷苯胺盐酸盐直接在1.0当量的碳酸钠的水溶液中与1.1当量的1,2-乙二硫醇反应4小时析出4-(1,3,2-二硫砷烷)苯胺,过滤,干燥。
上述步骤一中经过盐酸酸化成4-二氯砷苯胺盐酸盐后,经过过滤和干燥后再用于制备4-(1,3,2-二硫砷烷)苯胺;步骤一方法一中,4-二氯砷苯胺盐酸盐先在碱性条件下水解后,经过过滤、干燥,再在甲醇中与1,2-乙二硫醇反应,经过重结晶得到4-(1,3,2-二硫砷烷)苯胺;步骤一方法二中,4-二氯砷苯胺盐酸盐在碱性条件下直接与1,2-乙二硫醇反应,经过重结晶得到4-(1,3,2-二硫砷烷)苯胺。
上述步骤二的溶剂是二氯甲烷、四氢呋喃、N,N-二甲基甲酰胺、甲醇之一。反应温度是室温。步骤二反应完成后,经过柱层析得到N-(4-(1,3,2-二硫砷烷)苯基)炔酰胺。
上述步骤三中的溶剂是水与叔丁醇的混合溶剂(体积比为1:1)或者N,N-二甲基甲酰胺。在一价铜(抗坏血酸钠与二价铜盐如:硫酸铜、乙酸铜、三氟甲烷磺酸铜等原位生成即可或碘化亚铜)催化下发生点击反应,同时脱保护基生成N-(4”-氧化砷苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺。反应条件是在氮气保护的条件下,反应时间是100oC加热4小时或者微波100oC加热30min。步骤三的后处理经过硅藻土过滤,减压蒸馏,柱层析得到N-(4”-氧化砷苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺。
步骤四的反应条件是在甲醇中室温搅拌4小时。
步骤四反应完成后将反应体系浓缩,析出晶体或者柱层析,干燥得到N-(4”-(1”,3”,2”-二硫砷烷)苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺I。
本发明采用便宜的阿散酸为原料经过三步反应制备N-(4”-(1”,3”,2”-二硫砷烷)苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺I。
本发明所述化合物I具有抗直肠癌的活性,可以作为活性药物在制备抗直肠癌的药物中的应用。
本发明化合物I的制备路线:
具体实施方式
以下实例进一步说明本发明,并不意味着对本发明的限制:
实施例一:4-(1,3,2-二硫砷烷)苯胺的制备,甲醇作溶剂。通过方法一先制备4-二氯砷苯胺盐酸盐,再氨水作用下水解得到4-氨基苯基氧化砷(3价砷),再与1,2-乙二硫醇反应制备4-(1,3,2-二硫砷烷)苯胺。称取阿散酸(对氨基苯砷酸)(10g,46.08mmol),碘化钾(50mg,0.30)溶于60mL甲醇和48mL浓盐酸混合溶剂中。通入二氧化硫气体30min至溶液从橙红色变成浅黄色。白色固体析出。溶液降温至0oC,静置过夜。抽滤,滤饼用冷乙醚50mL洗涤两次。得到白色固体4-二氯砷苯胺盐酸盐(12.57g,95%)。
4-二氯砷苯胺盐酸盐(12.57g,43.78mmol)分批加入20mL浓氨水中,搅拌30min后,白色固体析出。过滤,滤饼用10mL水洗涤两次,真空干燥,得到白色固体4-氨基苯亚砷酸(3价)。
4-氨基苯亚砷酸(1.504g,7.675mmol)溶于150mL甲醇,室温下加入1,2-乙二硫醇(651uL,7.751mmol)反应4小时后,过滤,得到白色固体4-(1,3,2-二硫砷烷)苯胺(1.0g,收率50%)。
实施例二:4-(1,3,2-二硫砷烷)苯胺的制备,甲醇作溶剂。通过方法二先制备4-二氯砷苯胺盐酸盐,直接与1,2-乙二硫醇反应制备4-(1,3,2-二硫砷烷)苯胺。
称取阿散酸(对氨基苯砷酸)(10g,46.08mmol),碘化钾(50mg,0.30)溶于60mL甲醇和48mL浓盐酸混合溶剂中。通入二氧化硫气体30min至溶液从橙红色变成浅黄色。白色固体析出。溶液降温至0oC,静置过夜。抽滤,滤饼用冷乙醚50mL洗涤两次。得到白色固体4-二氯砷苯胺盐酸盐(12.57g,95%)。
4-二氯砷苯胺盐酸盐(12.57g,57.96mmol)溶于300mL水溶液中,冰水外降温至0-5oC,分批加入碳酸钠(共10.65g,57.96mmol),冰浴30min后加入1,2-乙二硫醇(4.86mL,57.96mmol)。搅拌4小时。析出白色固体4-(1,3,2-二硫砷烷)苯胺(9.68g,64%)。
实施例三:以4-(1,3,2-二硫砷烷)苯胺为原料,以二氯甲烷作溶剂。与炔丙酸在N,N’-环己基碳二亚胺作用下生成N-(4-(1,3,2-二硫砷烷)苯基)炔酰胺。
炔丙酸(216uL,3.521mmol)加入到溶有N,N’-环己基碳二亚胺(726.5mg,3.521mmol)的二氯甲烷(10mL)中,室温下搅拌1小时,冰浴降温搅拌30min后滴加(1,3,2-二硫砷烷)苯胺(760.6mg,2.934mmol)的二氯甲烷(10mL)。滴加完毕,保温反应30min,逐渐升温至室温反应过夜。反应完毕。硅藻土过滤,减压蒸干溶剂,残余物柱层析(淋洗液:石油醚:乙酸乙酯=4:1),得到黄色固体(584.5mg,64%)。
实施例四:2-叠氮-1,3,4,6-O-乙酰基-D-葡萄糖中间体与N-(4-(1,3,2-二硫砷烷)苯基)炔酰胺在叔丁醇与水(体积比为1:1)溶剂中在无水硫酸铜与抗坏血酸钠催化下发生点击反应生成N-(4”-氧化砷苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺。
2-叠氮-1,3,4,6-O-乙酰基-D-葡萄糖(70.5mg,0.1889mmol),N-(4-(1,3,2-二硫砷烷)苯基)炔酰胺(64.7mg,0.2079mmol),五水硫酸铜(106.3mg,0.4250mmol)和抗坏血酸钠(3.8mg,0.01889mmol)溶于1mLN,N-二甲基甲酰胺中,氮气保护下100oC反应4小时,反应完毕,减压蒸干溶剂。向残余物中加入5mL水和20mL乙酸乙酯萃取两次,合并有机相,无水硫酸钠干燥,过滤,减压蒸干。残余物经过柱层析(二氯甲烷:甲醇=20:1),得到白色固体N-(4”-氧化砷苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺(44mg,收率:38%)。
实施例五:N-(4”-氧化砷苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺在甲醇中与1,2-乙二硫醇反应生成产物N-(4”-(1”,3”,2”-二硫砷烷)苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺I。
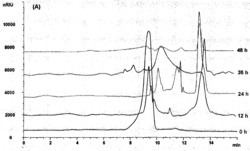
N-(4”-氧化砷苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺(44mg,0.07232mmol)溶于4mL甲醇,加入1,2-乙二硫醇(6.1uL,0.07305mmol)。室温下搅拌4小时,析出白色固体,重结晶得到白色晶体N-(4”-(1”,3”,2”-二硫砷烷)苯基)-1-(1’,3’,4’,6’-O-乙酰基-D-葡萄糖)-1,2,3-三唑-4-甲酰胺I(26mg,收率:52.55%)。产物为β和α构型的混合物。其中β:α=17:50经过1HNMR和13CNMR和HR-MS表征。
1HNMRα构型:δ=8.92(s,1H),8.28(s,1H),7.81-7.62(m,4H),6.42(d,J=3.2Hz,1H),6.06-6.01(m,1H),5.33-5.28(m,1H),4.72-4.67(m,1H),3.76(m,1H),3.39-3.32(m,2H),3.20-3.14(m,2H),2.14-1.99(m,12H);
1HNMRβ构型:δ=8.96(s,1H),8.23(s,1H),7.81-7.62(m,4H),6.28(d,J=8.4Hz,1H),5.92-5.87(m,1H),5.26-5.22(m,1H),4.42-4.35(m,1H),4.18-4.10(m,2H),3.48(m,1H),3.39-3.33(m,2H),3.20-3.14(m,2H),2.14-1.99(m,12H).
13CNMRα/β构型:δ=170.47,169.65,169.57,169.03,167.9,157.34,143.27,139.51,139.46,138.14,138.10,131.72,126.87,125.21,119.69,119.65,91.54,89.78,73.10,72.08,69.99,68.74,68.17,68.03,63.33,61.64,61.30,41.87,29.69,20.70,20.66,20.52,20.49,20.26,20.21;
HRMS:M+H+:C25H30AsN4O10S2理论值:685.0619,实测:685.0620.
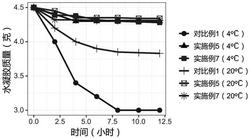
从下面实验可以看出化合物I所示的化合物具有良好的直肠癌生物活性。
实施例六:
实验方法
(1)细胞培养
结直肠癌细胞株HCT116由本实验室长期保存于液氮中,使用前培养于含10%胎牛血清的培养基RPMI培养基1640(GIBCO)中,置于37oC、饱和湿度及5%CO2培养箱常规培养,每2~4天传代,取对数生长期的细胞进行实验。
(2)MTS细胞毒性实验
取对数生长期的细胞结直肠癌细胞HCT116制备细胞悬液,接种在96孔细胞培养板中,在含有10%FBS的培养基中培养过夜,第二天加入不同浓度的化合物,在5%CO2培养箱培养;72h终止培养,加入MTS(Promega),37oC孵育1-4小时,在酶标仪(ThermoScientificVarioskanFlash)上于490nm读取吸光值。计算细胞毒性IC50。分别重复三次以上。取平均值。
化合物Ia的IC50=1.34uM,
实验表明:本发明中的I具有良好的抗直肠癌的活性,也可以用做肿瘤的抑制剂。
具有抗肿瘤活性的砷糖化合物及其制法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0