

IPC分类号 : B01J31/24,C07C2/36,C07C9/15,C07C9/16,C07F19/00,C07F15/04









专利摘要
本发明涉及一种烯烃齐聚催化剂及制备方法,以及其用于烯烃齐聚的方法。所述催化剂为采用膦配体和氮杂环卡宾配体双齿配位的镍氢化合物,增强了镍中心的稳定性,催化剂的热稳定性显著提高。适用于催化正丁烯或混合C4烯烃齐聚制备C8烯烃和C12烯烃。齐聚反应过程中不需要加入烷基铝为助催化剂,反应完后催化剂可循环套用,大大降低了催化剂的成本和三废的产生。
权利要求
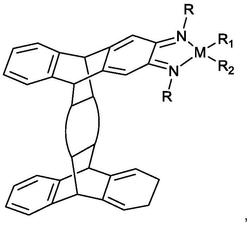
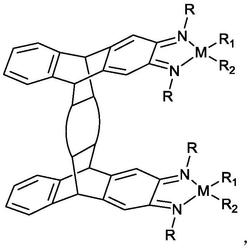
1.一种烯烃齐聚催化剂,所述催化剂的结构式为式I:
其中,L选自三苯基膦或三甲基膦;R选自C1~C10的烷基;Ph表示苯基;m、n相互独立的选自0或1。
2.根据权利要求1所述的催化剂,其特征在于,R选自甲基、乙基、丙基、丁基、正己基、正辛基或正癸基。
3.根据权利要求1所述的催化剂,其特征在于,所述催化剂选自结构式为式II、III的化合物:
其中,L选自三苯基膦或三甲基膦;R选自C1~C10的烷基;Ph表示苯基。
4.根据权利要求3所述的催化剂,其特征在于,R选自甲基、乙基、丙基、丁基、正己基、正辛基或正癸基。
5.根据权利要求1所述的催化剂,其特征在于,所述式I催化剂的制备方法,包括以下步骤:
(I-1)
(I-2)
(I-3)
其中X为Cl或Br。
6.根据权利要求3所述的催化剂,其特征在于,所述式II催化剂的制备方法,包括以下步骤:
(II-1)(2-溴苯基)二苯基膦与1-R-1H-咪唑反应制备
(II-2)
(II-3)
7.根据权利要求3所述的催化剂,其特征在于,所述式III催化剂的制备方法,包括以下步骤:
(III-1)
(III-2)
(III-3)
8.根据权利要求7所述的催化剂,其特征在于,所述化合物
9.一种烯烃齐聚的方法,包括以下步骤:正丁烯或混合C4烯烃在权利要求1-8任一项所述的催化剂的催化下,进行齐聚;所述烯烃包括正丁烯或混合C4烯烃,所述的混合C4烯烃包括以下组成,以混合C4烯烃的重量计:1-丁烯1~20wt%,顺-2-丁烯1~20wt%,反-2-丁烯1~10wt%,异丁烷30~50wt%,正丁烷20~50wt%。
10.根据权利要求9所述的方法,其特征在于,所述催化剂的用量为正丁烯或混合C4烯烃重量的0.01~2wt%。
11.根据权利要求10所述的方法,其特征在于,所述催化剂的用量为正丁烯或混合C4烯烃重量的0.1~1wt%。
12.根据权利要求9所述的方法,其特征在于,所述齐聚反应的温度20~80℃;反应压力为表压1~5MPa。
13.根据权利要求12所述的方法,其特征在于,所述齐聚反应的温度30~60℃;反应压力为表压2~4MPa。
说明书
技术领域
本发明涉及一种烯烃齐聚的催化剂及其制备方法,尤其涉及一种适用于正丁烯或混合C4烯烃齐聚的催化剂及其制备方法,还涉及正丁烯或混合C4烯烃齐聚的方法。
技术背景
异壬醇(Isononyl alcohol,简称INA),主要用于生产PVC类增塑剂邻苯二甲酸二异壬酯(DINP)。DINP是优良的通用型增塑剂,DINP与聚氯乙烯相容性好,挥发性、迁移性低,增塑效率高,性能优于邻苯二甲酸二辛酯(DOP)。DINP加氢制备的环己基二甲酸二异壬酯(DINCH),不含苯环,是真正的无毒环保型增塑剂。
异壬醇的生产工艺分为齐聚、氢甲酰化和加氢三步。目前齐聚工艺有三种:(1)采用镍盐为主催化剂,加入过量的烷基铝为助催化剂的齐格勒-纳塔催化剂体系催化正丁烯或混合C4烯烃齐聚,法国石油研究院(IFP)采用该工艺已经工业化,称为Dimersol工艺。该工艺的优点在于镍盐廉价易得,催化剂具有高活性和高选择性,但是体系为均相反应,催化剂无法循环套用,反应结束后需要使用酸或碱淬灭反应,产生大量废水,另外反应需要加入过量二氯乙基铝为助催化剂,铝镍摩尔比在几十至上百,烷基铝用量较大,催化剂成本大大增加,另外过量的烷基铝后处理时产生大量的废水,同时烷基铝易燃易爆,储存和使用的安全风险极大。(2)1983年赫斯化学和环球油品公司联合开发Octol工艺实现了工业化。该工艺采用固定床反应器,在一定条件下,丁烯-1转化率可达85%,辛烯选择性80%-83%,产物支链度较低,是用于生产增塑剂的优良原料。该工艺将硝酸镍负载于氧化铝上,高温焙烧,加入三氯化铝和二氯乙基铝活化。但是该催化剂仍需要使用大量的烷基铝用于催化剂制备,并且催化剂对水氧杂质敏感,极容易失活,产物中含有失活的烷基铝残留物,需要碱洗除去,产生大量废水。(3)1965年,法国石油研究院(IFP)开发的Difasol工艺是世界上第一个工业化的非均相丁烯齐聚工艺,该工艺使用镍基催化剂和烷基氯化铝助催化剂负载于离子液体上催化丁烯齐聚,反应过程中仍然不可避免的使用大量的烷基铝,并且使用离子液体为载体,成本昂贵,体系对水敏感,离子液体和催化剂均极容易分解,失活后难以再生。
上述催化体系均使用过量的烷基铝为助催化剂,显著增加了催化剂的成本和安全风险,并且有大量的三废产生,催化剂对水氧杂质敏感,失活后难以循环再生。
发明内容
本发明的目的在于提供一种烯烃齐聚的催化剂及其制备方法,所述催化剂尤其适用于催化正丁烯或混合C4烯烃齐聚,催化剂安全性高,热稳定性高,能够循环套用。
本发明还提供一种烯烃齐聚的方法,在所述催化剂的催化下,不使用烷基铝做助催化剂,原料转化率高,齐聚产品选择性高,三废减少,通过催化剂的循环套用,降低催化剂成本。
为了实现上述发明目的,本发明采用的技术方案如下:
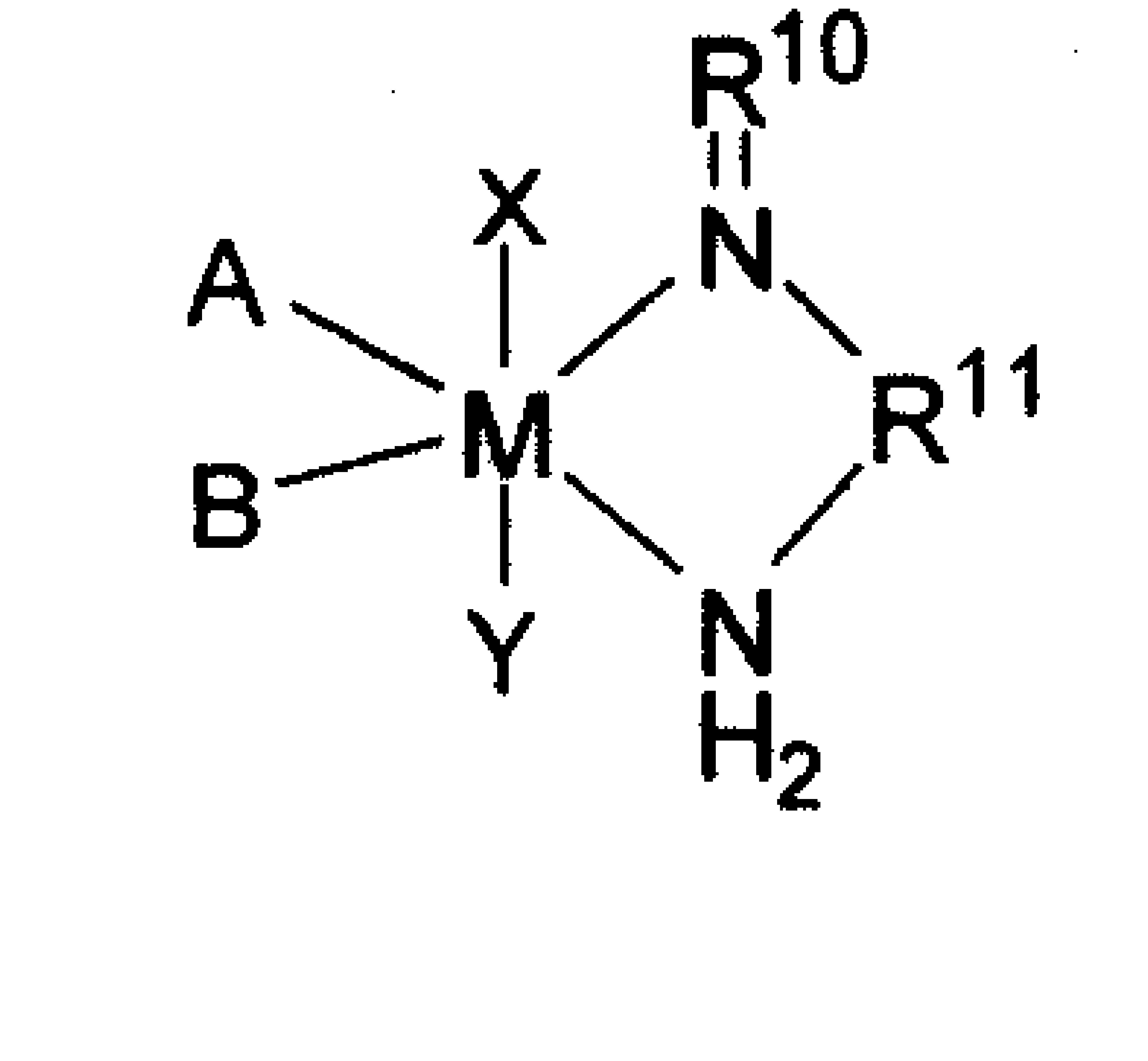
一种烯烃齐聚催化剂,所述催化剂的结构式为式I:
其中,L选自三苯基膦和/或三甲基膦;R选自C1~C10的烷基,优选甲基、乙基、丙基、丁基、正己基、正辛基或正癸基;Ph表示苯基;m、n相互独立的选自0或1。
优选的,催化剂选自结构式为式II、III的化合物:
其中,L选自三苯基膦和/或三甲基膦;R选自C1~C10的烷基,优选甲基、乙基、丙基、丁基、正己基、正辛基或正癸基;Ph表示苯基。
本发明所述式I的化合物的制备方法包括以下步骤:
(I-1) (I-a)与1-R-1H-咪唑反应制备 (I-b);
(I-2) (I-b)与NiCl2L2和t-BuOK反应制备 (I-c);
(I-3) (I-c)与LiAlH4反应制备 (I);
反应方程式如下:
其中X为Cl或Br。
本发明所述步骤(I-1)中,化合物I-a与1-R-1H-咪唑反应的温度为60~250℃,优选150~200℃,反应时间为1~4h,优选2~3h。降至室温,过滤得到化合物I-b。1-R-1H-咪唑与化合物I-a的摩尔比为1.1~1.2:1。步骤(I-1)可以在有或没有溶剂存在的条件下进行,所述溶剂优选己烷。
本发明所述步骤(I-2)中,化合物I-b与NiCl2L2和t-BuOK反应的温度为-20~20℃,优选-10~10℃,反应时间为1~4h,优选2~3h,结晶温度0~10℃,优选2~5℃。NiCl2L2与化合物I-b的摩尔比为1.1~1.2:1;t-BuOK与化合物I-b的摩尔比为1.2~1.5:1。步骤(I-2)在惰性气体优选N2气氛下进行。步骤(I-2)的反应介质为四氢呋喃,反应完毕后,除去四氢呋喃,加入己烷进行结晶,得到化合物I-c。
本发明所述步骤(I-3)中,化合物I-c与LiAlH4反应的温度为60~80℃,优选65~75℃,反应时间为2~4h,优选2.5~3.5h,结晶温度0~10℃,优选2~5℃。LiAlH4与化合物I-c的摩尔比为1.1~1.2:1。步骤(I-3)的反应介质为己烷。
优选的,本发明所述式II的化合物的制备方法包括以下步骤:
(II-1) ((2-溴苯基)二苯基膦,式II-a)与1-R-1H-咪唑反应制备 (II-b);
(II-2) (II-b)与NiCl2L2和t-BuOK反应制备 (II-c);
(II-3) (II-c)与LiAlH4反应制备 (II);
反应方程式如下:
本发明所述步骤(II-1)中(2-溴苯基)二苯基膦(II-a)与1-R-1H-咪唑的反应温度为60~250℃,优选150~200℃,反应时间为1~4h,优选2~3h,降温至室温后,使用二氯甲烷洗涤后,过滤得到化合物II-b。1-R-1H-咪唑与化合物II-a的摩尔比为1.1~1.2:1。
本发明所述步骤(II-2)中,化合物II-b与NiCl2L2、t-BuOK反应的温度为-20~20℃,优选-10~10℃,反应时间为1~4h,优选2~3h,结晶温度0~10℃,优选2~5℃。NiCl2L2与化合物I-b的摩尔比为1.1~1.2:1;t-BuOK与化合物II-b的摩尔比为1.2~1.5:1。步骤(II-2)在惰性气体优选N2气氛下进行。步骤(II-2)的反应介质为四氢呋喃,反应完毕后,除去(优选减压蒸馏)四氢呋喃,加入己烷进行结晶,得到化合物II-c。
本发明所述步骤(II-3)中,化合物II-c与LiAlH4反应的温度为60~80℃,优选65~75℃,反应时间为2~4h,优选2.5~3.5h,结晶温度0~10℃,优选2~5℃。LiAlH4与化合物II-c的摩尔比为1.1~1.2:1。步骤(II-3)的反应介质为己烷。反应完成后,降温至室温,过滤除去过量的LiAlH4,降温至0~10℃,放置过夜,结晶得到催化剂II。
优选的,本发明所述式III的化合物的制备方法包括以下步骤:
(III-1) (III-a)与1-R-1H-咪唑反应制备 (III-b);
(III-2) (III-b)与NiCl2L2和t-BuOK反应制备 (III-c);
(III-3) (III-c)与LiAlH4反应制备 (III);
反应方程式如下:
本发明所述步骤(III-1)中,化合物III-a与1-R-1H-咪唑反应的温度为60℃~250℃,优选150~200℃,反应时间为1~4h,优选2~3h,降至室温,过滤得到化合物III-b。1-R-1H-咪唑与化合物III-a的摩尔比为1.1~1.2:1。反应介质为己烷。
本发明所述步骤(III-2)中,化合物III-b与NiCl2L2、t-BuOK反应的温度为-20~20℃,优选-10~10℃,反应时间为1~4h,优选2~3h,,结晶温度0~10℃,优选2~5℃。NiCl2L2与化合物III-b的摩尔比为1.1~1.2:1;t-BuOK与化合物III-b的摩尔比为1.2~1.5:1。步骤(III-2)在惰性气体优选N2气氛下进行。步骤(III-2)的反应介质为四氢呋喃,反应完毕后,除去四氢呋喃,加入己烷进行结晶,得到化合物III-c。
本发明所述步骤(III-3)中,化合物III-c与LiAlH4反应的温度为60~80℃,优选65~75℃,反应时间2~4h,优选2.5~3.5h,结晶温度0~10℃,优选2~5℃。LiAlH4与化合物III-c的摩尔比为1.1~1.2:1。步骤(I-3)的反应介质为己烷。
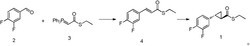
本发明所述化合物III-a可以采用以下方法制备,但不限于以下方法:邻二氯苄和丁基锂和二苯基氯化膦反应制备 (III-a);反应方程式如下:
优选的,本发明所述的制备化合物III-a的方法,包括以下步骤:温度为-78~-30℃,向邻二氯苄的己烷溶液中缓慢加入(优选滴加)丁基锂,然后缓慢升温至室温,继续反应1~2h,然后缓慢加入(优选滴加)二苯基氯化膦,继续反应2~4h,过滤得到化合物III-a;丁基锂与邻二氯苄的摩尔比为1.1~1.2:1,二苯基氯化膦与邻二氯苄的摩尔比为1.1~1.2:1。
本发明所述的烯烃齐聚催化剂可用于催化正丁烯或混合C4烯烃齐聚。所述的混合C4烯烃包括以下组成,以混合C4烯烃的重量计:1-丁烯1~20wt%,顺-2-丁烯1~20wt%,反-2-丁烯1~10wt%,异丁烷30~50wt%,正丁烷20~50wt%。
一种用本发明所述的催化剂催化正丁烯或混合C4烯烃齐聚的方法,包括以下步骤:正丁烯或混合C4烯烃在本发明所述的催化剂的催化下齐聚。
本发明所述催化剂的用量为正丁烯或混合C4烯烃重量的0.01~2wt%,优选0.1~1wt%;齐聚反应的温度20~80℃,优选30~60℃;反应压力为表压1~5Mpa,优选表压2~4MPa。
本发明所述的齐聚反应结束后,可以使用本领域公知的方法进行产品的分离与提纯。优选的,通过精馏分离二聚体C8烯和三聚体C12烯,剩余的含催化剂的四聚体溶液循环回齐聚反应,作为催化剂重复利用。
本发明所述的催化剂为采用膦配体和氮杂环卡宾配体双齿配位的镍氢化合物,增强了镍中心的稳定性,催化剂的热稳定性显著提高,精馏过程中可以保持稳定而不分解,因此催化剂的溶液可以直接循环回齐聚反应中,降低了催化剂的成本;同时由于膦的供电子作用和氮杂环卡宾配体的协同,镍氢化合物非常活泼,催化正丁烯或混合C4烯烃齐聚具有较高的活性。
以催化剂III为例,催化正丁烯齐聚的机理为:催化剂III与正丁烯发生配位后,镍氢插入形成中间体A,中间体A继续与正丁烯配位插入,生成中间体B,B继续发生正丁烯的配位和插入,生成中间体C,中间体B不稳定,发生消除反应生成催化剂III和正丁烯二聚体,中间体C不稳定发生消除反应,生成正丁烯三聚体和催化剂III,催化剂III继续循环反应。
反应机理如下式所示:
本发明与现有技术相比,有以下优点:
(1)直接以新型镍氢化合物为催化剂,不需要过量的烷基铝为助剂,无安全隐患,反应过程中无大量的三废产生。
(2)催化剂热稳定性高,可循环套用,大大降低了催化剂成本。
(3)催化剂活性和产物选择性明显升高,正丁烯或混合C4烯烃中的1-丁烯、顺-2-丁烯和反-2-丁烯的转化率可达到90%以上,产物二聚体选择性可达到90%以上,五次套用后转化率仍在90%以上,选择性85%以上。
具体实施方式:
下面结合实施例对本发明作进一步的详细说明,本发明的范围包括但不局限于此类实施例。
分析仪器及方法如下:
核磁:Varian-NMR 300,化学位移以ppm标示;
气相色谱-质谱联机(EI-MS):Finnigan MAT 95,70eV;
气相色谱仪:Agilent-7820;
气相色谱柱:0.25mm×30m的DB-5毛细管柱,检测器FID,气化室温度280℃,柱箱温度280℃,FID检测器温度300℃,氩气载流量2.1mL/min,氢气流量30mL/min,空气流量400mL/min,进样量1.0μL。使用面积归一化法计算烯的转化率及产物的选择性。升温程序:预热至柱温40℃,保持5min,15℃/min的速率从40℃升至280℃,保持2min。
实施例1
(1)催化剂的制备
(2-溴苯基)二苯基膦与1.1摩尔当量的甲基咪唑在60℃加热搅拌1h,降温至室温后,使用二氯甲烷洗涤三遍,过滤得到化合物1a,收率85%。
氮气保护下,化合物1a与1.1摩尔当量的NiCl2(PPh3)2、1.5摩尔当量的叔丁醇钾混合,加入所用反应物等质量四氢呋喃为溶剂,-20℃反应2h,反应完后,将四氢呋喃除去,加入己烷,过滤得到己烷溶液,降温至0℃,放置过夜,结晶析出固体,过滤得化合物1b,收率85%。1H NMR(300MHz,CDCl3):δ=3.81(s,3H),5.06(d,1H,J=10.5Hz),5.21(d,1H,J=10.5Hz),6.9-7.8(m,30H)。质谱分析数据:MS(EI)m/z(relative intensity):700,365,141,79。
化合物1b与1.1当量的LiAlH4混合,己烷为溶剂,60℃反应2h,降温至室温,过滤,降温至0℃,放置过夜,结晶析出催化剂1c,过滤得到产品,收率92%。1H NMR(300MHz,CDCl3):δ=-2.12(s,1H),3.81(s,3H),5.06(d,1H,J=10.5Hz),5.21(d,1H,J=10.5Hz),6.9-7.8(m,30H)质谱分析数据:MS(EI)m/z(relative intensity):701,365,141,79。
(2)正丁齐聚
正丁烯使用前4A分子筛干燥,水含量小于20ppm,反应釜中氮气保护下加入0.02g催化剂1c,加入200g正丁烯,氮气加压至表压1Mpa,反应温度20℃,反应时间2h,转化率95%,气相分析C8选择性91%,C12选择性6%,C16选择性3%。
(3)分离
闪蒸分离出正丁烯后,反应液连续精馏分离C8,塔板数20,气相色谱分析塔顶C8纯度99.2wt%,塔釜为C12和C16;连续精馏分离C12和C16,塔板数15,减压至绝对压力3Kpa,塔顶温度68-70℃收集C12,塔釜剩余C16及催化剂1c;塔釜液循环回反应釜继续反应。
(4)催化剂循环套用
塔釜液循环回反应釜,继续补加200g正丁烯反应,加压至表压2Mpa,反应温度40℃,反应2h,转化率92%,气相分析C8选择性90%,C12选择性5%,C16选择性5%;按照步骤(3)精馏分离产品,含催化剂的塔釜液循环返回反应釜,继续催化正丁烯齐聚,催化剂连续套用5次。
实施例2
(1)催化剂的制备
(2-溴苯基)二苯基膦与1.2摩尔当量的乙基咪唑250℃加热搅拌4h,降温至室温后,使用二氯甲烷洗涤三遍,过滤得到化合物2a,收率90%。1H NMR(300MHz,CDCl3):δ=1.29(t,3H,J=8.0Hz),4.12(q,2H,J=8.0Hz),7.71-7.89(m,16H),8.92(s,1H)质谱分析数据:MS(EI)m/z(relative intensity)439,359,174,79。
氮气保护下,化合物2a与1.2摩尔当量的NiCl2(PPh3)2、1.2摩尔当量的叔丁醇钾混合,加入所用反应物等质量四氢呋喃为溶剂,20℃反应4h,反应完后,将四氢呋喃除去,加入己烷,降温至10℃,放置过夜,析出固体,过滤得到化合物2b,收率87%。1H NMR(300MHz,CDCl3):δ=1.29(t,3H,J=8.0Hz),4.12(q,2H,J=8.0Hz),5.06(d,1H,J=10.5Hz),5.21(d,1H,J=10.5Hz),6.9-7.8(m,30H)质谱分析数据:MS(EI)m/z(relative intensity)715,360,175,79。
化合物2b与1.2当量的LiAlH4混合,己烷为溶剂,80℃反应4h,降温至室温,过滤,降温至10℃,放置过夜,结晶析出催化剂2c,过滤得到产物,收率93%。1H NMR(300MHz,CDCl3):δ=-2.51(s,1H),1.29(t,3H,J=8.0Hz),4.12(q,2H,J=8.0Hz),5.06(d,1H,J=10.5Hz),5.21(d,1H,J=10.5Hz),6.9-7.8(m,30H)质谱分析数据:MS(EI)m/z(relativeintensity)716,360,175,79。
(2)正定烯齐聚
正丁烯使用前4A分子筛干燥,水含量小于20ppm,反应釜中氮气保护下加入4g催化剂2c,加入200g正丁烯,氮气加压至表压5Mpa,反应温度80℃,反应0.5h,转化率98%,气相分析C8选择性94%,C12选择性4%,C16选择性2%。
(3)分离
闪蒸分离出正丁烯后,反应液连续精馏分离C8,塔板数20,气相色谱分析塔顶C8纯度99.5wt%,塔釜为C12和C16;连续精馏分离C12和C16,塔板数15,减压至绝对压力3Kpa,塔顶温度68-70℃收集C12,塔釜剩余C16及催化剂2c;塔釜液循环回反应釜继续反应。
(4)催化剂循环套用
塔釜液循环回反应釜,继续补加200g正丁烯反应,氮气加压至表压2Mpa,反应温度50℃,反应0.5h,转化率95%,气相分析C8选择性90%,C12选择性6%,C16选择性4%;按照步骤(3)精馏分离产品,含塔釜液的催化剂循环返回反应釜,继续催化正丁烯齐聚,催化剂连续套用5次。
实施例3
(1)催化剂的制备
(2-溴苯基)二苯基膦与1.2摩尔当量的正辛基咪唑150℃加热搅拌2h,降温至室温后,使用二氯甲烷洗涤三遍,过滤得到化合物3a,收率98%。1H NMR(300MHz,CDCl3):δ=0.88(t,3H,J=8.0Hz),1.29-1.74(m,12H,),4.04(d,2H,J=7.1Hz),7.3-8.1(m,16H),8.95(s,1H)。质谱分析数据:MS(EI)m/z(relative intensity)442,338,226,79。
氮气保护下,化合物3a与1.2摩尔当量的NiCl2(PPh3)2、1.2摩尔当量的叔丁醇钾混合,加入所用反应物等质量四氢呋喃为溶剂,-10℃反应2h,反应完后,将四氢呋喃除去,加入己烷,降温至2℃,放置过夜,析出固体,过滤得到化合物3b,收率90%。1H NMR(300MHz,CDCl3):δ=0.88(t,3H,J=8.0Hz),1.29-1.74(m,12H,),4.04(d,2H,J=7.1Hz),5.12(d,1H,J=8.0Hz),5.28(d,1H,J=10.5Hz),7.3-8.1(m,15H)。质谱分析数据:MS(EI)m/z(relative intensity)798,686,331,147,79。
化合物3b与1.2当量的LiAlH4混合,己烷为溶剂,65℃反应2.5h,降温至室温,过滤,降温至2℃,放置过夜,结晶析出催化剂3c,过滤得到产物,收率93%。1H NMR(300MHz,CDCl3):δ=-2.11(s,1H),0.88(t,3H,J=8.0Hz),1.29-1.74(m,12H,),4.04(d,2H,J=7.1Hz),5.12(d,1H,J=8.0Hz),5.28(d,1H,J=10.5Hz),7.3-8.1(m,15H)。质谱分析数据:MS(EI)m/z(relative intensity)799,687,331,147,79。
(2)正定烯齐聚
正丁烯使用前4A分子筛干燥,水含量小于20ppm,反应釜中氮气保护下加入0.2g催化剂3c,加入200g正丁烯,氮气加压至表压5Mpa,反应温度30℃,反应2h,转化率95%,气相分析C8选择性91%,C12选择性7%,C16选择性2%。
(3)分离
闪蒸分离出正丁烯后,反应液连续精馏分离C8,塔板数20,气相色谱分析塔顶C8纯度99.7wt%,塔釜为C12和C16;连续精馏分离C12和C16,塔板数15,减压至3Kpa,塔顶温度68-70℃收集C12,塔釜剩余C16及催化剂3c;塔釜液循环回反应釜继续反应。
(4)催化剂循环套用
塔釜液循环回反应釜,继续补加200g正丁烯反应,氮气加压至表压2Mpa,反应温度50℃,反应0.5h,转化率94%,气相分析C8选择性89%,C12选择性6%,C16选择性5%;按照步骤(3)精馏分离产品,含塔釜液的催化剂循环返回反应釜,继续催化正丁烯齐聚,催化剂连续套用5次。
实施例4
(1)催化剂的制备
(2-溴苯基)二苯基膦与1.2摩尔当量的正癸基咪唑200℃加热搅拌3h,降温至室温后,使用二氯甲烷洗涤三遍,过滤得到化合物4a,收率98%。
氮气保护下,化合物4a与1.2摩尔当量的NiCl2(PMe3)2、1.2摩尔当量的叔丁醇钾混合,加入所用反应物等质量四氢呋喃为溶剂,10℃反应3h,反应完后,将四氢呋喃除去,加入己烷,降温至5℃,放置过夜,析出固体,过滤得到化合物4b,收率92%。
化合物4b与1.2当量的LiAlH4混合,己烷为溶剂,75℃反应3.5h,降温至室温,过滤,降温至5℃,放置过夜,结晶析出催化剂4c,过滤得到产物,收率93%。
(2)正定烯齐聚
正丁烯使用前4A分子筛干燥,水含量小于20ppm,反应釜中氮气保护下加入2g催化剂4c,加入200g正丁烯,氮气加压至表压2Mpa,反应温度60℃,反应0.5h,转化率91%,气相分析C8选择性91%,C12选择性6%,C16选择性3%。
(3)分离
闪蒸分离出正丁烯后,反应液连续精馏分离C8,塔板数20,气相分析塔顶C8纯度99.3wt%,塔釜为C12和C16;连续精馏分离C12和C16,塔板数15,减压至3Kpa,塔顶温度68-70℃收集C12,塔釜剩余C16及催化剂4c;塔釜液循环回反应釜继续反应。
(4)催化剂循环套用
塔釜液循环回反应釜,继续补加200g正丁烯反应,氮气加压至表压2Mpa,反应温度50℃,反应0.5h,转化率90%,气相分析C8选择性91%,C12选择性7%,C16选择性2%;按照步骤(3)精馏分离产品,含塔釜液的催化剂循环返回反应釜,继续催化正丁烯齐聚,催化剂连续套用5次。
实施例5
(1)催化剂合成
-78℃,向30wt%邻二苄氯的己烷溶液中滴加1.1摩尔当量的丁基锂,滴加速度为2-5秒一滴,滴加完毕后缓慢升至室温,继续搅拌1h,将1.1摩尔当量的二苯基氯化膦滴加到上述溶液中,滴加完毕后搅拌2h,过滤得到化合物5a,收率83%。
室温下,化合物5a与1.1摩尔当量的丙基咪唑混合,己烷为溶剂,100℃反应2h,过滤得到化合物5b,收率88%。
氮气保护下,化合物5b与1.2摩尔当量的NiCl2(PPh3)2、1.2摩尔当量的叔丁醇钾混合,加入所用反应物等质量的四氢呋喃为溶剂,-15℃反应2h,反应完后,将四氢呋喃抽干,加入己烷,降温至8℃,放置过夜,析出固体,过滤得到化合物5c,收率85%。
化合物5c与1.2当量的LiAlH4混合,己烷为溶剂,70℃反应3h,降温至6℃,放置过夜,结晶析出催化剂5d,过滤得到产物,收率90%。
(2)正定烯齐聚
正丁烯使用前4A分子筛干燥,水含量小于20ppm,反应釜中氮气保护下加入1g催化剂5d,加入200g正丁烯,氮气加压至表压2Mpa,反应温度40℃,反应3h,转化率95%,气相分析C8选择性92%,C12选择性5%,C16选择性3%。
(3)分离
闪蒸分离出正丁烯后,反应液连续精馏分离C8,塔板数20,气相分析塔顶C8纯度99.3wt%,塔釜为C12和C16;连续精馏分离C12和C16,塔板数15,减压至绝对压力3Kpa,塔顶温度68-70℃收集C12,塔釜剩余C16及催化剂4e;塔釜液循环回反应釜继续反应。
(4)催化剂循环套用
塔釜液循环回反应釜,继续补加200g正丁烯反应,氮气加压至表压3Mpa,反应温度50℃,反应4.5h,转化率94%,气相分析C8选择性88%,C12选择性7%,C16选择性5%;按照步骤(3)精馏分离产品,含塔釜液的催化剂循环返回反应釜,继续催化正丁烯齐聚,催化剂连续套用5次。
实施例6
(1)催化剂合成
-30℃,向40wt%邻二苄氯的己烷溶液中滴加1.2摩尔当量的丁基锂,滴加速度为2-5秒一滴,滴加完毕后缓慢升至室温,继续搅拌2h,将1.2摩尔当量的二苯基氯化膦滴加到上述溶液中,滴加完毕后搅拌4h,过滤得到化合物5a,收率87%。
室温下,化合物5a与1.2摩尔当量1-丁基咪唑混合,己烷为溶剂,170℃反应2.5h,过滤得到化合物6a,收率90%。
氮气保护下,化合物6a与1.2摩尔当量的NiCl2(PPh3)2、1.5摩尔当量的叔丁醇钾混合,加入所用反应物等质量四氢呋喃为溶剂,5℃反应3h,反应完后,将四氢呋喃除去,加入己烷,降温至4℃,放置过夜,析出固体,过滤得到化合物6b,收率91%。
化合物6b与1.2当量的LiAlH4混合,己烷为溶剂,63℃反应3h,降温至1℃,放置3h,结晶析出催化剂6c,过滤得到产物6c,收率88%。
(2)正定烯齐聚
正丁烯使用前4A分子筛干燥,水含量小于20ppm,反应釜中氮气保护下加入0.5g催化剂6c,加入200g正丁烯,氮气加压至表压4Mpa,反应温度70℃,反应1.5h,转化率92%,气相分析C8选择性94%,C12选择性5%,C16选择性1%。
(3)分离
闪蒸分离出正丁烯后,反应液连续精馏分离C8,塔板数20,气相分析塔顶C8纯度99.9wt%,塔釜为C12和C16;连续精馏分离C12和C16,塔板数15,减压至绝对压力3Kpa,塔顶温度68-70℃收集C12,塔釜为C16及催化剂6c;塔釜液循环回反应釜继续反应。
(4)催化剂循环套用
塔釜液循环回反应釜,继续补加200g正丁烯反应,氮气加压至表压2Mpa,反应温度30℃,反应2h,转化率93%,气相分析C8选择性93%,C12选择性5%,C16选择性2%;按照步骤(3)精馏分离产品,含塔釜液的催化剂循环返回反应釜,继续催化正丁烯齐聚,催化剂连续套用5次。
实施例7
(1)催化剂合成
-60℃,向35wt%邻二苄氯的己烷溶液中滴加1.05摩尔当量的丁基锂,滴加速度为2-5秒一滴,滴加完毕后缓慢升至室温,继续搅拌1.5h,将1.1摩尔当量的二苯基氯化膦滴加到上述溶液中,滴加完毕后搅拌3h,过滤得到化合物5a,收率89%。
室温下,化合物5a与1.05摩尔当量正己基咪唑混合,己烷为溶剂,60℃反应2h,过滤得到化合物7a,收率88%。
478,442,358,277,79.
氮气保护下,化合物7a与1.2摩尔当量的NiCl2(PMe3)2、1.2摩尔当量的叔丁醇钾混合,加入所用反应物等质量四氢呋喃为溶剂,-10℃反应2h,反应完后,将四氢呋喃除去,加入己烷,降温至0℃,放置过夜,析出固体,过滤得到化合物7b,收率85%。
化合物7b与1.2当量的LiAlH4混合,己烷为溶剂,60℃反应2h,降温至0℃,放置过夜,结晶析出催化剂7c,过滤得到产物,收率90%。
(2)正定烯齐聚
正丁烯使用前4A分子筛干燥,水含量小于20ppm,反应釜中氮气保护下加入0.2g催化剂7c,加入200g正丁烯,氮气加压至表压2Mpa,反应温度40℃,反应0.2h,转化率91%,气相分析C8选择性91%,C12选择性7%,C16选择性2%。
(3)分离
闪蒸分离出正丁烯后,反应液连续精馏分离C8,塔板数20,气相分析塔顶C8纯度99.7wt%,塔釜为C12和C16;,连续精馏分离C12和C16,塔板数15,减压至绝对压力3Kpa,塔顶温度68-70℃收集C12,塔釜剩余C16及催化剂7c;塔釜液循环回反应釜继续反应。
(4)催化剂循环套用
塔釜液循环回反应釜,继续补加200g正丁烯反应,氮气加压至3Mpa,反应温度50℃,反应1h,转化率91%,气相分析C8选择性88%,C12选择性7%,C16选择性5%;按照步骤(3)精馏分离产品,含塔釜液的催化剂循环返回反应釜,继续催化正丁烯齐聚,催化剂连续套用5次。
实施例8
(1)催化剂合成
-40℃,向30wt%邻二苄氯的己烷溶液中滴加1.15摩尔当量的丁基锂,滴加速度为2-5秒一滴,滴加完毕后缓慢升至室温,继续搅拌1h,将1.1摩尔当量的二苯基氯化膦滴加到上述溶液中,滴加完毕后搅拌2h,过滤得到化合物5a,收率83%。
室温下,化合物5a与1.15摩尔当量正癸基咪唑混合,己烷为溶剂,60℃反应2h,过滤得到化合物8a,收率88%。
氮气保护下,化合物8a与1.2摩尔当量的NiCl2(PMe3)2、1.2摩尔当量的叔丁醇钾混合,加入反应物等质量的四氢呋喃为溶剂,-15℃反应2h,反应完后,将四氢呋喃除去,加入己烷,降温至0℃,放置过夜,析出固体,过滤得到化合物8b,收率85%。
化合物8b与1.2当量的LiAlH4混合,己烷为溶剂,60℃反应2h,降温至0℃,放置过夜,结晶析出催化剂8c,过滤得到产物,收率90%。
(2)正定烯齐聚
正丁烯使用前4A分子筛干燥,水含量小于20ppm,反应釜中氮气保护下加入0.2g催化剂8c,加入200g正丁烯,氮气加压至表压2Mpa,反应温度40℃,反应0.2h,转化率91%,气相分析C8选择性91%,C12选择性7%,C16选择性2%。
(3)分离
闪蒸分离出正丁烯后,反应液连续精馏分离C8,塔板数20,气相分析塔顶C8纯度99.5wt%,塔釜为C12和C16;,连续精馏分离C12和C16,塔板数15,减压至绝对压力3Kpa,塔顶温度68-70℃收集C12,塔釜剩余C16及催化剂8c;塔釜液循环回反应釜继续反应。
(4)催化剂循环套用
塔釜液循环回反应釜,继续补加200g正丁烯反应,氮气加压至表压3Mpa,反应温度50℃,反应1h,转化率91%,气相分析C8选择性92%,C12选择性7%,C16选择性1%;按照步骤(3)精馏分离产品,含塔釜液的催化剂循环返回反应釜,继续催化正丁烯齐聚,催化剂连续套用5次。
实施例9
(1)混合C4齐聚
混合C4使用前4A分子筛干燥,水含量小于20ppm,组成为1-丁烯9wt%,顺2-丁烯11wt%,反2-丁烯6wt%,异丁烷44wt%,正丁烷30wt%。反应釜中氮气保护下加入1.4g催化剂8c,加入200g正丁烯,氮气加压至表压2Mpa,反应温度40℃,反应3h,转化率93%,气相分析C8选择性92%,C12选择性6%,C16选择性2%。
(2)分离
闪蒸分离出未反应的C4,连续精馏分离C8,塔板数20,气相分析塔顶C8纯度99.6wt%,塔釜为C12和C16;连续精馏分离C12和C16,塔板数15,减压至绝对压力3Kpa,塔顶温度68-70℃收集C12,塔釜C16及催化剂4;塔釜液循环回反应釜继续反应。
(3)催化剂循环套用
塔釜液循环回反应釜,继续补加200g正丁烯反应,氮气加压至表压2Mpa,反应温度40℃,反应2h,转化率93%,气相分析C8选择性92%,C12选择性6%,C16选择性2%;按照步骤(3)精馏分离产品,含塔釜液的催化剂循环返回反应釜,继续催化正丁烯齐聚,催化剂连续套用5次。
实施例1-9的结果见表1。
表1实施例1-9结果
对比例
正丁烯使用前4A分子筛干燥,水含量小于20ppm,反应釜中氮气保护下加入1.4g辛酸镍和15ml二氯乙基铝,搅拌反应10分钟后,加入200g正丁烯,氮气加压至表压2Mpa,反应温度40℃,反应3h,转化率78%,气相分析C8选择性85%,C12选择性10%,C16选择性5%。
一种烯烃齐聚催化剂及制备方法,以及其用于烯烃齐聚的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0