

IPC分类号 : B01J21/00,B01J23/755,B01J35/04,B01J37/02,B01J37/12,B01J37/18,B01D53/94,B01J23/889,B01J35/00,C23C8/00







专利摘要
用于特别是从碳发电设备的废气中同时除去NOx和碳颗粒的整料催化剂包含由耐酸奥氏体钢制成的整料。催化剂是两相催化剂;而且,该整料催化剂包含具有尖晶石结构的NiFe2O4和Fe2O3(Mh)相,且这些相形成微晶,还包含Mn。用于特别是从碳发电设备的废气中同时除去NOx和碳颗粒的整料催化剂的制备方法取决于,使整料进行氧化,之后将所得的氧化物层用镍盐溶液洗涤;之后将整料在氧化气氛中烘烤以将镍离子插入到氧化物层中;最后使该所得的氧化物层进行还原。
权利要求
1.一种用于同时除去NO
其中所述整料由耐酸奥氏体钢制成,且所述催化剂是两相催化剂;而且,所述整料催化剂包含具有尖晶石结构的NiFe
2.根据权利要求1所述的方法,其特征在于,所述整料催化剂用于从碳发电设备的废气中同时除去NO
3.根据权利要求1所述的方法,其特征在于,所述整料的氧化在空气流中进行。
4.根据权利要求1或2所述的方法,其特征在于,使所述整料在使用热程序对其加热时进行氧化。
5.根据权利要求1或2所述的方法,其特征在于,使所述整料在其加热至700~800℃的温度范围时进行氧化,之后将其在最终温度下烘烤。
6.根据权利要求5所述的方法,其特征在于,使所述整料在最终温度下氧化2~6小时。
7.根据权利要求1或2所述的方法,其特征在于,将所氧化的整料的氧化物层用浓度为0.1~0.25mol/dm
8.根据权利要求7所述的方法,其特征在于,将所述氧化物层用镍离子溶液洗涤的操作通过将所述整料浸在可溶的镍盐溶液中进行。
9.根据权利要求8所述的方法,其特征在于,将所述整料浸在所述镍盐溶液中1~4秒。
10.根据权利要求7所述的方法,其特征在于,将乙酸镍溶液用于洗涤所述氧化物层。
11.根据权利要求1或2所述的方法,其特征在于,在将所述氧化物层用镍离子溶液洗涤之后,使用热程序使所述整料进行另一加热至700~800℃的温度并在最终温度下烘烤2小时。
12.根据权利要求1或2所述的方法,其特征在于,将所述氧化物层用所插入的镍进行还原的操作在氢流中进行。
13.根据权利要求12所述的方法,其特征在于,氢流速度取决于所述整料的体积。
14.根据权利要求12所述的方法,其特征在于,将所述氧化物层用所插入的镍离子进行还原的操作在使用热程序将所述整料加热至400~500℃的最终温度的同时进行。
15.根据权利要求14所述的方法,其特征在于,使在最终温度下的还原进行0.5~1.5小时。
16.根据权利要求12所述的方法,其特征在于,将所还原的催化剂在氢流中冷却至室温。
说明书
技术领域
本发明的主题是在耐酸钢基底上的同时除去NOx和碳颗粒的整料(monolithic)催化剂,特别是从碳发电设备的废气中同时除去NOx和碳颗粒。通常将称为去NOx的技术用于从静止的排放源例如发电设备、CHP设备、废物焚化设备或硝酸工厂除去氮氧化物。该技术的基础是,在基于放置于锐钛矿晶型TiO2上的五氧化二钒的催化剂上,用氨还原氮氧化物。除五氧化二钒(含量为低于1%)外,当前用在该技术中的催化剂通常还包含显著量的三氧化钨(ca.10%)或三氧化钼(ca.6%)(Appl.Catal.222(2001)221-236,P.Forzatti)。三氧化物增加该催化剂的活性和热稳定性,并且它们降低引起硫酸铵沉积的形成的SO2到SO3的氧化。除此之外,MoO3防止由砷化合物(如果它们存在于排气中的话)引起的催化剂失活(Chemiker Zeitung 2(1991)33,E.Hums)。
背景技术
近来有关于基于五氧化二钒的催化剂的专利申请公开指出合成方法的有利改变或促进剂的量和质的改变的可能性(例如,公开WO 2013002492(A1))。申请US 2011250114(A1)还公开了三氧化钼和三氧化钨以及降低三氧化钼挥发性的组分的同时使用。
去NOx技术中的在陶瓷整料上的氧化物V-W催化剂的表面可能同时被正在生成的硫酸铵以及经过静电沉淀器的烟灰小颗粒堵塞。而且,氨是较为昂贵的还原剂,其需要费钱的分配装置且还污染空气,因而对人的呼吸系统具有不利影响。
可以将催化剂放置在陶瓷整料通道的表面上和/或孔中。根据波兰专利PL 199013B1的描述,其被放置在金属整料通道的表面上。然而,陶瓷载体很脆弱,且其壁很厚。
因此,理想的解决方案是基于金属箔的耐用的整料催化剂。
另一方面,在高度吸热过程中由氧和氮形成的氮氧化物包含高内能。因此,使用合适的催化剂应当容易将其还原成氧和氮。该氧化物颗粒的电子结构示出,其最低未占轨道是反键π轨道。因而,吸附NO颗粒的表面氧化物阳离子的电子越丰富(还原程度越大),吸附于表面氧化物阳离子的NO颗粒的解离更容易(Catal.Today 73(2002)249-254,M.Najbar等人)。因为在静止排放源的废气中存在2~7%的氧,氧化物催化剂表面的高还原状态的保持仅可以在催化剂的操作温度高到足以保持气相中的氧压低于氧对氧化物的平衡压力的情况下实现,不然则需要使用额外的还原剂。
在含有碳颗粒的多尘土的废气中,在能够进行氧化物表面还原的温度下可以发生NO的直接还原而无需引入额外的还原剂。很有可能的是,在碳氧化物下发生还原,该碳氧化物在先前的于碳颗粒上的NO还原过程中形成和/或因颗粒与形成在氧化物表面的原子形式氧的反应而形成(J.Phys.Chem.B 103(1999)3434T.Kyotani&A.Tomita)。因而,预期在氧化物上的O2离解以及由氧化物上离解的氧活化的碳颗粒上的NO分解同时发生的温度范围内会发生对NO的最优分解。
在US 20060073964(A1)中,以及在US 20070025901中,氧载体上的贵金属被描述为用于在含氧气氛中直接分解氮氧化物而无需额外还原剂的催化剂。
另一方面,在EP 0526099A1中,以及在EP 0679427A1中,氧化物载体上的银或银氧化物被描述为用于在含氧气氛中直接分解氮氧化物而无需额外还原剂的催化剂,并且在KR101011830(B1)中,这些催化剂是水滑石结构的含有过渡金属的金属氧化物。
提出将具有尖晶石结构的混合氧化物催化剂用于在还原因子的存在下的直接NO分解(JP 7284663、JP 10180105)。
在混合的V-W氧化物催化剂上观察到在低于氧离解温度的温度下的直接NO分解(PP 2006-344611)。
使用αFe2O3上活化氧的在烟灰上的直接分解NO的测试记载于多个公开中(AppliedCatalysis B:Environmental 80(2008)248-259D.Reichert等人、Applied Catalysis B:Environmental 84(2008)803-812D.Reichert等人)。
同时检定在金属铁上的无氧情况下的NO分解(Chemical Engineering Science,53(1998)1481-1489,A.N.Hayhurst等人和Fuel 89(2010)3505-3509,B.Gradoń等人)。
不幸的是,上述催化剂均未能以工业规模使用。
发明内容
本发明的目的是开发整料催化剂的合成,该整料催化剂包含整料钢结构上的氧化物相,其特征在于其在直接NO分解中由高活性引起的还原状态以及在废气所含碳颗粒的存在下对于氮的较高且稳定的选择性。
根据本发明,用于特别是从碳发电设备的废气中同时除去NOx和碳颗粒的整料催化剂包含由耐酸钢制得的整料(monolith),该整料通过交替放置的正弦波状箔带和光滑箔带形成。交替放置的带优选使用“双S法”进行折叠并且它们在圆柱体箱中形成整体的圆柱体块(cubes)和/或它们可以放置在立方体箱中,从而形成整体的立方体块。
优选地,应当使用具有以下组成的钢:2.0%Mn、0.8%Si、0.045%P、0.03%S、0.3%Cu、19.0%Cr、10.0%Ni和0.8%Ti。最优选的选项为奥氏体钢1H18N9T/1.4541。
催化剂的氧层位于整料通道的表面上,包含具有尖晶石结构的相(NiFe2O4和FeCr2O4);这些相形成微晶,还包含Mn。催化剂的合适氧化物的比表面积优选为1~5m
根据本解决方案,从碳发电设备的废气中同时除去NOx和碳颗粒的整料催化剂的制备方法在于通过交替放置正弦波状且光滑的箔的带来形成整料。交替放置的带使用“双S法”折叠并放置在圆柱体箱中,从而形成整体的圆柱体块,或者它们可以放置在立方体箱中,从而形成整体的立方体块。优选地,使用具有以下组成的钢:2.0%Mn、0.8%Si、0.045%P、0.03%S、0.3%Cu、19.0%Cr、10.0%Ni和最终的0.8%Ti。最优选的选项为奥氏体钢H18N9T/1.4541或1H18N9/1.4541。
通过在以2~6度/分钟的速度加热至高达700~800℃的温度的时间内进行氧化,然后在该温度下在含氧气氛中烘烤2~6小时,直接在由耐酸钢制成的整料上产生氧化物相。在整料以炉冷却速度冷却至室温后,将整料通道的表面上形成的氧层用镍盐溶液通过将整料浸在其中而洗涤。在整料在空气中于室温干燥2~6小时后,将其在空气流中使用热程序再次加热至高达700~800℃的温度,并且将其在最终温度下烘烤2~6小时。在炉冷却至室温后,通过在1个小时内以200~300cm
优选地,整料在以90~150cm
优选地,整料在高至730~750℃的温度的空气流中烘烤。
优选地,整料的形状和尺寸与炉腔的形状和尺寸严格匹配。
优选地,乙酸镍溶液用于洗涤氧化物层。
优选地,整料浸在镍盐溶液中的时间为1~4秒。
优选地,溶液中镍离子的浓度为0.1~0.25摩尔/dm
优选地,还原温度为500℃。
作为耐酸钢烘烤过程的结果,主要的氧化物层形成在钢通道的表面上,其主要暴露于磁铁矿和赤铁矿,但也很可能暴露于锰铁氧体。因洗涤主要层以及接下来对箔进行氧化而形成的氧化物层中的主相是镍铁氧体NiFe2O4的尖晶石相以及具有尖晶石结构的称为meghemite(Mh)的Fe2O3相的混合物。Cr、Mn和Ti原子溶在这些相中。使用氢的还原使铬、锰和钛扩散到氧化物层的微晶中,引起铁和镍的表面浓度的增加。
基于耐酸钢的尖晶石催化剂的特征为,在相对低温(300~350℃)下在碳发电设备的废气中在同时分解NO和氧化碳颗粒方面的较好活性。
在整料通道壁上形成的根据本发明的厚度为0.1~0.4μm的氧化物催化剂的薄致密层不被切下,且它们对它们下方的金属表面提供较好的针对进一步腐蚀的保护。
所提出的在耐酸钢制成的整料上的氧化物催化剂的其他益处是其合成的简单和相对低的成本。
催化剂表面的形态和活性氧层的厚度已经使用扫描电镜(SEM)并结合X射线能量分散谱(EDS)进行了检定。
表面氧化物层的相组成已经使用拉曼光谱法进行了检定。表面氧化物纳米层的化学组成已经使用X射线光电子谱(XPS)进行了定义。
直接NO分解的最佳速度在于氧化物表面上预期到最大浓度的原子氧种类且同时碳因吸附于氧化物表面的原子氧种类而被氧化的温度下实现。在整料通道的壁上形成的氧化物催化剂层(Mn-Fe-Cr-Ni-Ti)以较高的表面还原暴露尖晶石相,这促进电子传递到吸附的电子受体颗粒。所提出的基于耐酸钢制成的箔的整料催化剂在表面上暴露尖晶石相,主要包含镍铁氧体NiFe2O4和铬酸铁(FeCr2O4)以及氧化锰。这些相形成微晶,其还另外包含Mn。
所提出的在耐酸钢制成的整料上的Cr-Mn-Fe-Ni-Ti氧化物催化剂的其他益处为其合成的简单和在其中使用的基底的相对低成本。
本发明的主题已经在以下执行实施例中呈现。
具体实施方式
将三个由耐酸1H18N9T/1.4541钢制成的箔条使用热程序以3°/min的速度加热至740℃的温度。它们在最终温度下烘烤4个小时。在炉冷却后,对其中一箔条进行保护以用于进一步研究,且将剩余两个箔条上的氧层用0.1M的Ni(CH3COO)2溶液通过浸在溶液中2秒而进行洗涤。在将箔条干燥后,使用热程序在空气流中再次加热至740℃的温度,并在该温度下烘烤4个小时。在炉于空气流中冷却后(在约4个小时后),对其中一个箔条进行保护以用于进一步测试。将留在炉内的箔条上的氧化物层进行还原。为此,首先,通过以200cm
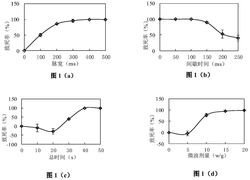
所得的在:氧化膜(O)(a)、在用乙酸镍溶液洗涤主要氧层之后再氧化的膜(OP)(b)、以及之后用氢还原的该再氧化膜(OPR(c))上的氧层的拉曼光谱呈现在图1中。
以下相的峰位置已经在最大光谱值上方标出:赤铁矿αFe2O3(Phys.Rev.B 41(1990)7822-7827M.J.Massem等人)(225、290、300、415、506、613和1320cm
正如可以容易观察到的,O箔的氧化物层的主相为赤铁矿和磁铁矿。除少量的maghemite外,具有NiFe2O4网络参数的尖晶石相在OP箔的氧化物中为主导。镍铁氧体的形成表明在盐微晶的表面层中的高镍含量,这由镍的强偏析(低Tamman温度)及其在主要氧化物层形成中的低参与(低氧亲和性)造成。OPR箔上的氧化物层的主相也是镍铁氧体。在OP箔上氧化物相的情况下,峰位置与观察到的位置的较小差异归因于由还原诱导的氧化物相中的扩散过程。(使用扫描电镜并结合X射线能量分散谱的催化剂研究结果提供对其表面的形态以及氧化物层厚度的信息)。图2呈现出在OP箔表面上的氧化物层的二次电子(SE)的SEM图像。氧化物相的结合微晶紧紧地覆盖箔的晶粒的表面。该箔的微区域的EDS(能量分散谱)分析使得能够使用Anderson-Hasler公式估算氧化物层的厚度(X-ray Optics andMicroanalysis,1966,310Herman编著,Paris C.Anderson,M.Hasler),等于约0.3μm。
氧化物层的表面纳米层的分析使用XPS法进行。
表1列出对于铁、铬、锰、钛和镍的2p电子结合能量以及这些元素在所分析的氧化物纳米层中的相对百分份额。
表1.在厚度为0.05mm的1H18N9T钢箔上的2p电子结合能和不同金属在表面氧化物纳米层中的百分比份额。
正如可以容易观察到的,表面氧化物纳米层的组成强烈取决于其先前的热处理。在O箔的表面纳米层中,具有更高氧亲和性的元素Cr、Mn和Ti的浓度大大高于初始箔(Cr-6原子%;Mn-2原子%;Ti 0.2原子%)的表面纳米层中的浓度(Adv.Mat.Res.651(2013)319E.Bielańska等人),这表明其由氧化诱导的表面偏析。存在于OP和OPR箔上的在NiFe2O4和Fe2O3-磁赤铁矿微晶的表面纳米层中的高含量Cr、Mn和Ti也表明它们由氧化诱导的强表面偏析,这因经尖晶石结构的扩散而发生。尖晶石结构中锰的阳离子取代先前通过透射电镜(TEM)并结合EDS和电子衍射在具有相似组成的粉末催化剂中观察到(专利申请P.395905)。
氧化物层的还原以所有金属的2p电子结合能的降低来表现,并且伴随有具有最高氧亲和性的金属(Cr、Mr和Ti)浓度的略微降低。
测试根据实施例1获得的两相催化剂(NiFe2O4–Fe2O3(Mh))在碳发电设备的废气气氛中同时分解NO和氧化碳颗粒的活性。使用风扇将排气引导至包含5个直径为9.8cm、高度为3cm且体积为226cm
测试使用来自碳焚化炉的排气通道的旁路来进行。在250~450℃的范围内进行NOx浓度的测定,在测定过程中,在各个指定温度(250、300、350、400和450℃)下的NOx浓度在200~400ppm的范围内变化。同时,SO2浓度在160~250ppm的范围内变化。将便携式HORIBAPG-250气体分析仪用于NOx、SO2、O2和CO2浓度的测定。测定在5150~19250h
图3示出NOx转化的温度依赖性。
正如可以容易观察到的,NOx分解在300和350℃的温度下最快发生。在350℃温度下其随时间的稳定性呈现在图4中。在源自NO分解的氧的作用下的去活化的缺失表明其用于沉积在催化剂的表面上的碳纳米颗粒的氧化。
图5呈现出时间追溯中400℃下废气与催化剂之间的相互作用的影响下的CO2和O2浓度的变化(△(CO2)和△(O2))。在第一个169小时内观察到的约2%的CO2浓度增加伴随着约2%的O2浓度降低(△(CO2)=-△(O2)),这表明碳颗粒到二氧化碳的催化氧化。在超过169小时的时间段中,碳颗粒氧化的速度首先增加,之后开始二氧化碳到碳和氧的离解。这表明,催化剂表面的逐渐演变发生在400℃,这引起也能够离解CO2的强电子供体中心在那个表面上的生成。催化剂表面在较高温度下的逐渐演变可能是其在NOx分解中较低活性的原因。这类催化剂表面的演变没有在300和350℃的温度下观察到。
因此,可以得出结论,在300~350℃的温度下,在本发明中记载的催化剂可以同时用于从碳发电设备的废气中除去NOx和碳纳米颗粒。
如Kyotani和Tomita所主张的(J.Phys.Chem.B 103(1999)3434T.Kyotani&A.Tomita),吸附于氧化物相的原子形式氧扩散到碳颗粒的表面上,这引起碳原子表面部分的氧化。这继而引起剩余C原子周边这样的改变,使得它们转变为NO离解的活性中心。同时,形成在碳表面的CO从氧化物表面除去过量的活性氧,从而暴露金属原子,其可变为NO离解的活性中心。氧化物相与碳颗粒之间的协同反应似乎是造成在300℃观察到(图3)最高NOx转化值的原因。基于Kyotani和Tomita工作的上述解决方案不是对于NO分解中氧化物和碳相协同的唯一解释。碳颗粒的活化氧化也可以在NO2下发生,NO2因NO分子与吸附于氧化物表面的原子氧的反应而形成(Appl.Catal..B 92(2009)126I.Atribak等人)。
特别是从碳发电设备的废气中同时除去NOx和碳颗粒的整料催化剂及其制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



















动态评分
0.0