










专利摘要
一种用密度泛函理论研究DBDS与DBS对铜绕组腐蚀性能的方法,首先利用分子模拟软件建立Cu(110)表面、Cu(110)与DBDS复合模型以及Cu(110)与DBS复合模型,其次对复合模型进行结构优化,可以得到复合模型的功函变化,然后计算DBDS与DBS在Cu(110)表面上的吸附能,最后计算并分析DBDS分子与DBS分子的前线轨道分布、前线轨道能量差、电负性以及电荷分布。本发明提出一种用密度泛函理论研究DBDS与DBS对铜绕组腐蚀性能的方法。与现在常用的实验方法相比,具有无损耗、节约成本、简单易行、可重复性高等优点。
权利要求
1.一种用密度泛函理论分析DBDS与DBS对铜绕组腐蚀性能的方法,其特征在于,包括以下步骤:
步骤1:在分子模拟软件内建立Cu(110)表面、Cu(110)/DBDS复合模型以及Cu(110)/DBS复合模型;
步骤2:基于密度泛函理论的平面波超软赝势对步骤1中的Cu(110)/DBDS复合模型以及Cu(110)/DBS复合模型进行结构优化,优化完成后可以得到Cu(110)/DBDS和Cu(110)/DBS的功函变化;
步骤3:在步骤2的结构优化的基础上,对Cu(110)/DBDS复合模型以及Cu(110)/DBS复合模型进行能量优化,完成后计算DBDS与DBS的吸附能;
步骤4:以DND为基组,泛函数设为广义梯度近似GGA,对DBDS分子与DBS分子进行结构优化,在结构优化后的模型上进行能量优化,计算得到前线轨道分布和前线轨道能量值,分析DBDS分子与DBS分子的稳定性;
步骤5:在Cu(110)/DBDS体系和Cu(110)/DBS体系结构和能量优化的基础上,对电荷分布进行分析,分析DBDS分子与DBS分子与铜的反应活性,即可完成DBDS与DBS对铜绕组腐蚀性能的分析。
2.根据权利要求1所述的用密度泛函理论分析DBDS与DBS对铜绕组腐蚀性能的方法,其特征在于,所述的功函变化的计算公式如式(1)、式(2)所示:
ΔΦ
ΔΦ
式(1)中Φ
ΔΦ
3.根据权利要求1所述的用密度泛函理论分析DBDS与DBS对铜绕组腐蚀性能的方法,其特征在于,所述的步骤2中,所述的结构优化的条件为收敛精度控制为2×10
4.根据权利要求1所述的用密度泛函理论分析DBDS与DBS对铜绕组腐蚀性能的方法,其特征在于,所述的步骤3中,计算吸附能如式(3)、式(4)所示:
E
E
其中E
E
E
E
5.根据权利要求1所述的用密度泛函理论分析DBDS与DBS对铜绕组腐蚀性能的方法,其特征在于,所述的步骤4中,采用所述的前线轨道分布得出两前线轨道能量差以及电负性;所述的两前线轨道能量差为ΔE=E
说明书
技术领域
本发明一种用密度泛函理论分析DBDS与DBS对铜绕组腐蚀性能的方法,涉及电气设备绝缘老化领域。
背景技术
电力变压器在保障电网安全运行上起着至关重要的作用,而油纸绝缘系统性能的好坏决定了电力变压器的使用寿命。二十一世纪以来,国内外报道了数起因变压器油中含有腐蚀性硫化物而引发的绝缘事故,对故障设备进行解体研究,分析在铜绕组及绝缘纸表面的沉积物,得出了硫化亚铜是引发设备故障的主要原因。
日本三菱公司提出了Cu2S在变压器内部的生成机理,指出首先油中的二苄基二硫醚(Dibenzyl-disulfide,DBDS)与铜离子结合形成DBDS-Cu复合物,其次DBDS-Cu复合物分解形成二苄基硫醚(Dibenzyl-sulfide,DBS)、二苯乙烷(Benzyl-1,2,BiBZ)以及Cu2S,其中DBS可再与铜反应生成Cu2S和BiBZ。从Cu2S在变压器内部的生成机理可以看出,DBDS与DBS都能与Cu反应并生成Cu2S,即都能腐蚀铜绕组。
分析出DBDS与DBS两种腐蚀性硫化物,哪种对变压器内铜绕组的腐蚀性能更强,有利于针对性的进行脱硫处理、选用合适的铜缓蚀剂,保护变压器的铜绕组,提高变压器的绝缘性能。目前此方面的研究大多数是以实验的手段进行,对微观机理的探索需进一步研究。
发明内容
本发明需要解决的问题是:针对研究DBDS与DBS对铜绕组的腐蚀缺乏微观的探索,提出一种用密度泛函理论研究DBDS与DBS对铜绕组腐蚀性能的方法。与现在常用的实验方法相比,具有无损耗、节约成本、简单易行等优点。
本发明采取的技术方案:
首先利用分子模拟软件建立Cu(110)表面、Cu(110)与DBDS复合模型以及Cu(110)与DBS复合模型,其次对复合模型进行结构优化,可以得到复合模型的功函变化,然后计算DBDS与DBS在Cu(110)表面上的吸附能,最后计算并分析DBDS分子与DBS分子的前线轨道分布、前线轨道能量差、电负性以及电荷分布。通过对上述情况具体分析,可以得到DBS容易吸附在铜表面,而DBDS更容易与铜发生反应。
步骤1:在分子模拟软件内建立Cu(110)表面、Cu(110)/DBDS复合模型以及Cu(110)/DBS复合模型;
步骤2:在CASTEP模块中基于密度泛函理论的平面波超软赝势对步骤1中的Cu(110)/DBDS复合模型以及Cu(110)/DBS复合模型进行结构优化,优化完成后可以得到Cu(110)/DBDS和Cu(110)/DBS的功函变化,功函变化的计算公式如式(1)、式(2)所示:
ΔΦ1=ΦDBDS+Cu(110)-ΦCu(110)
(1)
ΔΦ2=ΦDBS+Cu(110)-ΦCu(110)(2)
式(1)中ΦDBDS+Cu(110)表示DBDS/Cu(110)体系的功函,ΦCu(110)代表Cu(110)表面的功函数;式(2)中ΦDBS+Cu(110)表示DBS/Cu(110)体系的功函,ΦCu(110)代表Cu(110)表面的功函数;
ΔΦ1、ΔΦ2分别为Cu(110)/DBDS和Cu(110)/DBS的功函变化;
步骤3:在步骤2的结构优化的基础上,对Cu(110)/DBDS复合模型以及Cu(110)/DBS复合模型进行能量优化,完成后计算DBDS与DBS的吸附能,计算吸附能如式(3)、式(4)所示:
Eads1=EDBDS+Cu(110)-EDBDS-ECu(110)
(3)
Eads2=EDBS+Cu(110)-EDBS-ECu(110)
(4)
其中EDBDS+Cu(110)为吸附分子DBDS与Cu(110)表面的总能量;
EDBS+Cu(110)代表DBS与Cu(110)表面的总能量;
EDBDS、EDBS是分别将DBDS、DBS分子放入一个 的立方盒子计算出来的能量;ECu(110)代表纯Cu(110)表面的能量;
Eads1、Eads2为DBDS与DBS的吸附能;
步骤4:在DMol3模块中对DBDS分子与DBS分子进行结构优化,计算精度Quality设为Medium,基组选为DND,DND为双数值轨道基组+d轨道极化函数。与DN基组类似,但是为所有非氢原子加入了d轨道函数极化。为DMol3模块默认基组,在可接受的计算时内,能够确保精度在可接受的范围内。也是DMol3模块计算的最低可信精度基组。采用电子分布热平滑加快结构优化收敛,泛函数设为广义梯度近似GGA,在结构优化后的模型上进行能量优化,计算完成后得到前线轨道分布和前线轨道能量值,分析DBDS分子与DBS分子的稳定性;其中泛函数广义梯度近似是为克服局域密度近似存在的缺点而引入的,它在前者的基础上引入了对粒子数密度的梯度修正,大幅度减少了局域密度近似的误差,成为广义梯度近似。与局域密度近似相比,广义梯度近似全面的考虑到了每个区间内电子数密度对交换关联能的贡献和相邻区间的不同电子数密度对交换关联能的贡献,更适合处理具有非均匀密度的电子体系。
步骤5:在Cu(110)/DBDS体系和Cu(110)/DBS体系结构和能量优化的基础上,对电荷分布进行分析,分析DBDS分子与DBS分子与铜的反应活性,即可完成DBS与DBS对铜绕组腐蚀性能的分析。
所述的步骤2中,所述的结构优化的条件为收敛精度控制为2×10
所述的步骤3中,所述的能量优化的条件为收敛精度控制为2×10
所述的步骤4中,采用所述的前线轨道分布得出两前线轨道能量差以及电负性;所述的两前线轨道能量差为ΔE=ELUMO-EHOMO,其中,ELUMO为DBDS与DBS的最低未占轨道能量;EHOMO为DBDS与DBS的最高占据轨道能量;所述的电负性χ可由下式表示:χ=(I+A)/2,式中I=-EHOMO;A=-ELUMO。
本发明一种用密度泛函理论研究DBDS与DBS对铜绕组腐蚀性能的方法,技术效果如下:
1:本发明深入DBDS、DBS、Cu材料内部,利用密度泛函理论,分子模拟方法分析功函变化、吸附能、前线轨道分布、前线轨道能量差、电荷分布等,从微观层面探索DBDS与DBS的腐蚀性能的差异性。
2:与现在常用的实验方法相比,具有无损耗、节约成本、简单易行、可重复性高
等优点。对从微观层面探索腐蚀性硫化物的腐蚀性能提供微观信息。
附图说明
图1为Cu晶胞与Cu(110)晶面图,其中,a为Cu晶胞,b为Cu(110)晶面图。
图2为Cu(110)/DBDS复合模型、Cu(110)/DBS复合模型图,其中,a为Cu(110)/DBDS复合模型、b为Cu(110)/DBS复合模型图。
图3为DBDS分子、DBS分子、BiBZ分子模型图。
图4为Cu(110)表面静电势、Cu(110)/DBDS体系静电势、Cu(110)/DBS体系静电势图。
图5为DBDS分子与DBS分子前线轨道分布图。
图6为优化后的Cu(110)/DBDS复合模型与Cu(110)/DBS复合模型图,其中,a为优化后的Cu(110)/DBDS复合模型,b为Cu(110)/DBS复合模型图。
具体实施方式
一种用密度泛函理论研究DBDS与DBS对铜绕组腐蚀性能的方法,首先利用分子模拟软件建立Cu(110)表面、Cu(110)与DBDS复合模型以及Cu(110)与DBS复合模型,其次对复合模型进行结构优化,可以得到复合模型的功函变化,然后计算DBDS与DBS在Cu(110)表面上的吸附能,最后计算并分析DBDS分子与DBS分子的前线轨道分布、前线轨道能量差、电负性以及电荷分布。
一种用密度泛函理论研究DBDS与DBS对铜绕组腐蚀性能的方法,包括以下步骤:
步骤1:在分子模拟软件内,导出Cu晶胞模型,将Cu晶胞模型进行结构优化并进行真空层设置和切面处理,模型如图1所示。
步骤2:采用广义梯度近似GGA中的PBE方法,基于密度泛函理论的平面波超软赝势对DBDS/Cu(110)以及DBS/Cu(110)复合模型进行结构优化,优化选取的收敛精度为2×10
ΔΦ1=ΦDBDS+Cu(110)-ΦCu(110)
(1)
ΔΦ2=ΦDBS+Cu(110)-ΦCu(110)(2)
式(1)中ΦDBDS+Cu(110)表示DBDS/Cu(110)体系的功函,ΦCu(110)代表Cu(110)表面的功函数;式(2)中ΦDBS+Cu(110)表示DBS/Cu(110)体系的功函,ΦCu(110)代表Cu(110)表面的功函数。ΔΦ1、ΔΦ2若为负,则说明有效电荷从Cu(110)表面转移到了表面上方区域,即在表面和吸附物之间的区域电荷密度增加。静电势图如图4所示,从图4可以看出,DBDS/Cu(110)体系功函大小为4.11eV;DBS/Cu(110)体系功函大小为3.37eV;Cu(110)表面功函数大小是4.498eV。根据式(1)、式(2)可知ΔΦ1为-0.388eV,ΔΦ2为-1.118eV。由于S原子电负性(2.58)高于Cu原子电负性(1.9),在DBDS/Cu、DBS/Cu界面处存在电荷密度的重排,降低了Cu(110)表面的功函数,从而导致ΔΦ1、ΔΦ2为负值。|ΔΦ1|<|ΔΦ2|,说明DBS在Cu(110)表面的吸附性强于DBDS。
步骤3:表征分子吸附能力强弱的一个指标是分子与表面结合强度的大小,可以通过吸附能(Eads)来描述。在结构优化的基础上,对复合模型进行能量优化,能量优化完成可以计算DBDS与DBS的吸附能,计算吸附能如式(3)、式(4)所示:
Eads1=EDBDS+Cu(110)-EDBDS-ECu(110)
(3)
Eads2=EDBS+Cu(110)-EDBS-ECu(110)
(4)
其中EDBDS+Cu(110)为吸附分子DBDS与Cu(110)表面的总能量;EDBS+Cu(110)代表DBS与Cu(110)表面的总能量;EDBDS、EDBS指的是分别将DBDS、DBS分子放入一个
的立方盒子计算出来的能量;ECu(110)代表纯Cu(110)表面的能量。根据定义,Eads为负值时,表示吸附过程是自发的,是一个放热的过程,若Eads为正值时,表示吸附过程不是自发的,需要从外界吸取能量才能完成吸附过程,是一个吸热的过程。各能量变化如表1所示。由表1可知,Eads1大小为8.571eV,Eads2大小为6.077eV,Eads1、Eads2均大于0,说明DBDS与DBS不能自发地在铜表面吸附,需要从外界吸收能量才能吸附。DBDS需要从外界吸收8.571eV的能量进行继续吸附,而DBS需要从外界吸收6.077eV的能量。从吸附能来看,也是DBS更容易吸附在Cu(110)表面。根据Cu2S在变压器内部的生成机理,可知DBDS与DBS都能与Cu发生反应形成新的物质,伴随化学键的断裂和生成,此时DBDS与DBS在Cu(110)表面的吸附属于化学吸附。
表1 DBDS/Cu(110)和DBS/Cu(110)体系吸附能(eV)
DBDS分子模型以及DBS分子模型如图2所示,在DMol3模块中对DBDS分子与DBS分子进行结构优化,计算精度Quality设为Medium,基组选为DND,DND为双数值轨道基组+d轨道极化函数。与DN基组类似,但是为所有非氢原子加入了d轨道函数极化。为DMol3模块默认基组,在可接受的计算时内,能够确保精度在可接受的范围内。也是DMol3模块计算的最低可信精度基组。采用电子分布热平滑加快结构优化收敛,泛函数设为广义梯度近似GGA。在结构优化后的基础上,对图2的模型进行能量优化,能量优化的积分计算精度为fine,自洽场收敛精度为Medium,泛函数选择GGA,基组为DND,在性质选项中勾选Orbital选项,Orbital为轨道选项,计算完成后可得到前线轨道分布和前线轨道能量值,分析DBDS分子与DBS分子的稳定性。其中泛函数广义梯度近似是为克服局域密度近似存在的缺点而引入的,它在前者的基础上引入了对粒子数密度的梯度修正,大幅度减少了局域密度近似的误差,成为广义梯度近似。与局域密度近似相比,广义梯度近似全面的考虑到了每个区间内电子数密度对交换关联能的贡献和相邻区间的不同电子数密度对交换关联能的贡献,更适合处理具有非均匀密度的电子体系。
DBDS分子与DBS分子前线轨道分布如图5所示,从图5可以看出DBDS分子和DBS分子的HOMO轨道主要集中在S原子附近,DBS的LUMO轨道主要分布在苯环上,而DBDS的LUMO轨道除了分布在苯环上,在C-S键及S-S键也有分布,说明DBDS分子接受Cu表面电子的区域更加广泛。此外,两个前线轨道能量差ΔE能衡量分子的稳定性,ΔE数值越大,分子越稳定;ΔE数值越小,分子越不稳定,越容易发生化学反应
χ=(I+A)/2(5)
式中I=-EHOMO;A=-ELUMO,ΔE、χ以及EHOMO、ELUMO汇总如表2所示。
表2 DBDS及DBS分子特性汇总
由表2可知,DBDS的电负性大小为3.132eV,DBS的电负性大小为3.100eV,两者相差0.032eV,基本相等,说明DBDS和DBS分子束缚电子的能力基本相同。而DBDS分子的前线轨道能量差为2.610eV,DBS分子前线轨道能量差为3.610eV,两者有1eV的差距,说明DBDS分子的稳定性要小于DBS分子,从ΔE可以得出DBDS分子活性更大,不稳定性更高,DBDS更容易与Cu发生反应。
步骤5:在Cu(110)/DBDS体系和Cu(110)/DBS体系结构和能量优化的基础上,对其电荷分布进行分析。能量优化完成后获取了DBDS/Cu(110)体系中DBDS的电荷分布,同理也得到了DBS的电荷分布,由于原子数较多,限于表格篇幅,选取了H原子电荷总量QH、S原子电荷总量QS、C原子电荷总量QC作为分析对象,如表3所示。DBDS中C-S键、S-S键的变化,DBS中C-S键变化如表4所示。
表3 DBDS与DBS电荷分布
表4 C-S键和S-S键的变化
表3中Total指的是该分子得失电荷后的总电荷量,负为得到电荷,正为失去电荷。从总体上看,DBDS分子从铜表面上获得了1.99e电荷,DBS分子从Cu表面得到了1.65e电荷。由于DBDS分子电负性(3.132eV)略大于DBS分子电负性(3.100eV),DBDS得到的电荷量稍大于DBS,与电负性分析的结果一致。从电荷转移可知,DBDS中C和S所得到的4.70e电荷来源于H原子向C和S提供的2.71e电荷以及Cu表面向C和S提供的1.99e电荷;同理,DBS中C原子得到了4.41e电荷,一部分来源于H原子提供的2.56e电荷,另一部分由S原子提供0.20e,其余部分是由Cu表面提供的1.65e。可以看到DBDS的S原子是得电子,而DBS分子中的S原子失去电子,而铜表面带正电,说明DBDS分子的S原子更容易与Cu结合,如图6(a)所示。从图6(a)可以看出DBDS的两个S原子已到苯环下方,更接近Cu表面;而图6(b)中的S原子没有明显向Cu靠近的趋势。从表4中可知优化前S-S键长为 优化后的S-S键长为 说明此时S-S键有断裂的趋势;而DBS分子在DBS/Cu(110)体系优化前的C-S键长为 优化后的C-S键长为 优化前后键长变化不明显。表明DBS的稳定性高于DBDS,DBDS更容易与Cu发生反应,生成Cu2S、DBS以及其他副产物,其中Cu2S能严重破坏变压器的绝缘性能。
一种分析DBDS与DBS对铜绕组腐蚀性能的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
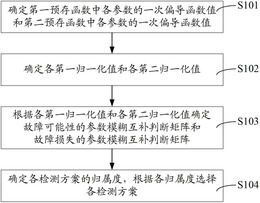
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0