







专利摘要
本发明涉及一种通过紫外分光光度法准确测定壳聚糖含量的方法,属于大分子检测领域。本发明利用壳聚糖在醋酸‑醋酸钠缓冲溶液中,与萘酚绿B、吐温20溶液反应生成缔合物,在一定的浓度范围内,其吸光度值随壳聚糖含量的增加而增大,并且呈现一定的线性关系。以此作为壳聚糖的定量基础,建立了测定壳聚糖的紫外分光光度法,本方法受壳聚糖的分子量的影响,具有试剂廉价,灵敏度高,重现性好和操作简便等优点。
权利要求
1.一种通过紫外分光光度法准确测定壳聚糖含量的方法,其包括下述步骤:
1)绘制ΔA和不同分子量的壳聚糖浓度的标准曲线:向10mL比色管中依次加入1.0mL一定浓度梯度的低分子壳聚糖标准溶液,1.5mL浓度为2.0×10
2)10μg/mL的样品工作液的制备:称取一定量待检测样品的胶囊,使用冰醋酸溶解并将其定容得到样品储备液,用漏斗脱脂棉过滤储备液,滤液经6000r/min,离心机离心20min,取上清液2.5mL于100mL容量瓶,定容,得浓度为10μg/mL的样品工作液;
3)根据壳聚糖分子量选择标准曲线:使用步骤1)中检测方法绘制ΔA和样品工作液的标准曲线,并将其与不同分子量的壳聚糖标准曲线比较,确定相对应的壳聚糖的分子量所使用的回归方程;
4)样品中壳聚糖含量确定:取样品工作液1mL,按步骤1)检测方法进行测定,在478nm处测定其吸光度值A,并计算ΔA=A-A0,将ΔA代入线性回归方程求得样品中壳聚糖的含量,同时做加标回收试验;
所述的醋酸-醋酸钠缓冲溶液pH为4.50;步骤1)中溶液的加入顺序为首先加入壳聚糖标准溶液,其次加入萘酚绿B,再者加入吐温20表面活性剂溶液,最后加入醋酸-醋酸钠缓冲液;所述浓度为6.0×10-2g/L吐温20表面活性剂溶液加入量为1.50ml;冷却至室温至测定的时间为8min-2h;步骤1)标准曲线C1,C2与C3中壳聚糖的浓度范围1.0-4.0μg/mL。
说明书
技术领域
本发明涉及一种通过紫外分光光度法准确测定壳聚糖含量的方法,属于大分 子检测领域。
背景技术
壳聚糖(chitosan)为甲壳素的降解产物。化学名称为聚葡萄糖胺(1,4)-2-氨基 -2-脱氧-D-葡聚糖,是甲壳类(虾、蟹)动物、昆虫的外骨骼的主要成分,壳聚 糖在自然界中分布广泛,属于高分子绿色材料。有关人类对壳聚糖的认识是从19 世纪开始的,到本世纪60年代,对甲壳素/壳聚糖的研究才变得日益活跃。国际 上,从1894年以来,已召开过6次有关甲壳素/壳聚糖的会议;亚洲也于1994 年举办了首届甲壳素学术会议,现已举办2次。而我国只是在最近几年才对甲壳 素/壳聚糖的开发及应用予以重视。1996年10月在大连召开了第一届甲壳素化学 学术会议。壳聚糖及其衍生物在食品、医药、环保、化工等行业有着广阔的应用 前景。壳聚糖具有很好的生物相容性和生物活性,无毒、无害、无免疫抗原性, 对人体具有强化免疫、抑制老化、预防疾病、促使疾病痊愈、调节生理机能五大 功能。市面上的壳聚糖产品参差不齐,配方不一,壳聚糖所占的含量直接影响到 其功能的发挥。因此,建立准确的定量分析方法,对保障产品质量的稳定性和可 控性都至关重要。
目前,壳聚糖分析研究及测定的方法主要有分光光度法、荧光分析法、电化 学法、红外光谱法测、液相色谱法、气相色谱法等。如韩美娜]等人研究建立荧 光淬灭法测定壳聚糖凝胶膜中壳聚糖的含量,利用壳聚糖在PBS缓冲溶液中对 异硫氰酸荧光素(FITC)具有明显的荧光淬灭作用,且荧光淬灭程度与壳聚糖 的量在一定范围内呈线性关系,从而建立了荧光淬灭法测定壳聚糖含量。商军和 王蓓两人建立高效液相色谱法测定原料和复合预混料中的低聚壳聚糖含量。谭学 才,麦智彬等人建立了以茜素红(AR)为电活性探针间接测定无电活性的壳聚糖 (壳聚糖)的新方法。由于壳聚糖自身容易降解,且紫外吸收较弱,实验中常采用 酸水解间接比色法进行含量分析,此方法中常常用到浓硫酸,操作具有一定的危险性,并且不适合微量壳聚糖含量的分析检测。气相色谱法测定壳聚糖含量时往 往需要在测定前对壳聚糖进行衍生化处理,操作较为复杂。HPLC法用于壳聚糖 含量测定时通常也是先将壳聚糖进行酸水解后再进行测定。这些酸水解或衍生化 操作过程对壳聚糖含量的测定结果都会有较大影响,一些直接测定壳聚糖含量的 方法逐渐受到关注。
基于此,本发明提供一种通过紫外分光光度法准确测定壳聚糖含量的方法。
发明内容
为了克服现有技术中对于壳聚糖的含量难以定量,操作技术繁琐的技术不足, 本发明提供一种通过紫外分光光度法准确测定壳聚糖含量的方法,本发明利用 壳聚糖在醋酸-醋酸钠缓冲溶液中,与萘酚绿B、吐温20溶液反应生成缔合物, 在一定的浓度范围内,其吸光度值随壳聚糖含量的增加而增大,并且呈现一定的 线性关系。以此作为壳聚糖的定量基础,建立了测定壳聚糖的紫外分光光度法, 本方法受壳聚糖的分子量的影响,具有试剂廉价,灵敏度高,重现性好和操作简 便等优点,适合在实际应用中推广使用。
一种通过紫外分光光度法准确测定壳聚糖含量的方法,其包括下述步骤:
1)绘制ΔA和不同分子量的壳聚糖浓度的标准曲线:向10mL比色管中依次 加入1.0mL一定浓度梯度的低分子壳聚糖标准溶液,1.5mL浓度为2.0×10
2)10μg/mL的样品工作液的制备:称取一定量待检测样品的胶囊,使用冰 醋酸溶解并将其定容得到样品储备液,用漏斗脱脂棉过滤储备液,滤液经
6000r/min,离心机离心20min,取上清液2.5mL于100mL容量瓶,定容,得浓 度为10μg/mL的样品工作液。
3)根据壳聚糖分子量选择标准曲线:使用步骤1)种检测方法绘制ΔA和样 品工作液的标准曲线,并将其与不同分子量的壳聚糖标准曲线比较,确定相对应 的壳聚糖的分子量所使用的回归方程。
4)样品中壳聚糖含量确定:取样品工作液1mL,按步骤1)检测方法进行 测定,在478nm处测定其吸光度值A,并计算ΔA=A-A0,将ΔA代入线性回归 方程求得样品中壳聚糖的含量,同时做加标回收试验。
如上所述的通过紫外分光光度法准确测定壳聚糖含量的方法,所述的醋酸- 醋酸钠缓冲溶液pH为4.50。
如上所述的通过紫外分光光度法准确测定壳聚糖含量的方法,步骤1中溶液 的加入顺序为首先加入壳聚糖标准溶液,其次加入萘酚绿B,再者加入吐温20 表面活性剂溶液,最后加入醋酸-醋酸钠缓冲液。
如上所述的通过紫外分光光度法准确测定壳聚糖含量的方法,所述浓度为 6.0×10
如上所述的通过紫外分光光度法准确测定壳聚糖含量的方法,冷却至室温至 测定的时间为8min-2h。
如上所述的通过紫外分光光度法准确测定壳聚糖含量的方法,步骤1)标准 曲线C1,C2与C3中壳聚糖的浓度范围1.0-4.0μg/mL。
样品前处理:将待测胶囊去胶囊壳,称取一定量样品,用冰醋酸溶解,定容 得样品储备液。用漏斗脱脂棉过滤储备液,滤液经6000r/min,离心机离心20min, 取上清液2.5mL于100mL容量瓶,定容,得浓度为10μg/mL的样品工作液。
样品测定:用样品工作液按实验方法绘制样品曲线,与不同分子量的壳聚糖 标准曲线比较,根据样品的回归方程确定样品中的壳聚糖的分子量的大小,根据 所对应的壳聚糖的分子量的大小确定所使用的ΔA和壳聚糖浓度的标准曲线;
取样品工作液1mL,在478nm处测定其吸光度值A,并计算ΔA=A0-A,将 ΔA代入线性回归方程求得样品中壳聚糖的含量,同时做加标回收试验
实验例:
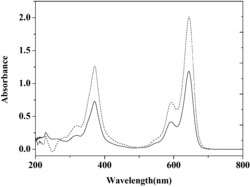
1、体系的紫外分光光谱图
图1为体系的紫外吸收曲线图。自上而下CTS浓度依次降低及CTS,吐温 20和萘酚绿B自身吸光度。由图1可知,CTS,吐温20和萘酚绿B本身的吸光 度值非常低,但当CTS与萘酚绿B及吐温20结合后吸光度值明显增大,并在 478nm处的线性关系是最好的,实验的空白组散射值较低,且实验组随着CTS 的浓度的增大,体系的散射值增大,存在线性关系。
2、缓冲体系及pH
在pH3.0~6.0范围内分别考察甘氨酸-盐酸、B-R缓冲溶液、醋酸-醋酸钠 HAc-NaAc三种缓冲溶液对体系吸光度的影响。结果表明,萘酚绿B与壳聚糖的 反应在HAc-NaAc缓冲溶液中灵敏度较大。如图2所示,根据HAc-NaAc缓冲溶 液的酸度对该反应体系的影响可以看出,缓冲溶液的酸度对该反应体系有着显著 的影响,pH4.50时体系的灵敏度较大且稳定,故选择HAc-NaAc缓冲溶液pH4.50。
3、缓冲溶液加入量的影响
缓冲溶液为实验体系提供合适的酸度结合环境,其加入量对离子缔合物的形 成有着一定的影响,不改变其他实验条件,改变pH=3.0的醋酸-醋酸钠缓冲溶液 的加入量为1.0,1.5,2.0,3.0,4.0mL,测定其吸光度值。测定结果见图3。绘 制ΔA-V曲线,结果表明,最佳缓冲溶液的用量是1.50ml。
4、萘酚绿B加入量的影响
萘酚绿B作为染料探针与目标物质壳聚糖结合,其加入量直接影响着离子 缔合物的形成,不改变其他实验条件,改变浓度为2.0×10
5、增敏剂的选择以及用量的影响
某些表面活性剂能起到一定增敏作用,故本试验考察了SDS、SLS、OP-10、 聚乙烯醇、吐温20、吐温80、SDBS对体系吸光度的影响。结果如图5所示, 萘酚绿B与壳聚糖的反应在吐温20溶液中增敏效果最好,。
其他条件不变,对吐温20按1.00,1.50,2.00,2.50,3.00,3.50mL加入体系中, 测量吸光度值,绘制ΔA-V曲线。如图6所示,吐温20的用量对该体系影响表 明,最佳的吐温20的用量为1.50ml时,灵敏度最高。故选择吐温20的用量为 1.50ml。
6、加入顺序的影响
在实验条件下,如表1所示,考察了萘酚绿B,醋酸-醋酸钠缓冲液,CTS 及吐温20的12种不同的加入顺序对体系灵敏度的影响。结果见图7,最佳加入 顺序为“CTS+萘酚绿B+吐温20+醋酸-醋酸钠缓冲液”,此时体系的吸光度最大, 灵敏度最高且重现性最好。
表1加入顺序的影响
7、反应温度及加热时间的影响
在实验条件下,考察了50~90℃不同温度对体系灵敏度的影响,结果如图8, 温度对体系的吸光度值存在着影响。吸光度值随着温度的升高呈上升趋势,80℃ 达到最大灵敏度且在该反应温度下,重现性较好,80℃后呈下降趋势,故选择反 应温度为80℃。
在此温度的优化条件下,考察了加热时间对该体系的影响,由实验结果表明, 当CTS-NGB体系在80℃加热8min时,体系的灵敏度ΔA最大且最稳定。因此,选 择该体系的加热条件为80℃水浴8min。
8、反应时间和稳定时间的影响
根据实验条件,反应结束后冷却至室温。前半小时每隔10min测定吸光度值, 半小时后以每隔20min测定。结果可见,该体系的吸光度在10min内即可达到 稳定,2h内基本不发生变化,稳定性良好。
9、离子强度的影响
实验中,用NaCl(0~0.30mol/L)考察了对该反应的吸光度值的影响。结果 表明(如图9),ΔA随NaCl浓度变化的总体趋势是先减后缓,在0-0.05mol/L时 内反应趋于平稳呈负影响,可能原因是NaCl与NGB竞争CTS使得ΔA下降。
10、共存物质的影响
考察了20种共存物质对该体系的干扰,体系中壳聚糖的浓度为1μg/mL。在 相对误差在±5%范围内,各种共存物质的允许量见表2。常见的氨基酸,糖类, 阳离子Na
表2共存物质的影响
11、线性范围与检出限
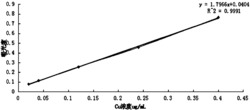
在最佳实验条件下,按照实验方法测定不同浓度的壳聚糖对应的ΔA,绘制 标准曲线,结果表明,壳聚糖在浓度1.0-4.0μg/mL范围内与ΔA存在良好的线性 关系,线性回归方程为y=0.0142x-0.0011,相关系数0.9974,方法线性范围宽。 根据公式:DL=3S/K,全部代入得DL=0.2470μg/mL。
12、不同分子量的壳聚糖结果比较
在最佳的实验条件下,考察该体系对不同分子量的壳聚糖的测定结果差异。 分别考察了低分子量、中分子量和高分子量的壳聚糖。经过统计学分析,不同分 子量的壳聚糖结果存在显著差异性,该方法测定壳聚糖含量受不同分子量的影响。
表3不同分子量的壳聚糖结果
本发明与现有技术相比具有如下技术优势:
1)线性好、检出限低:在最佳实验条件下,按照实验方法测定不同浓度的 壳聚糖对应的ΔA,绘制标准曲线,结果表明,壳聚糖在浓度1.0-4.0μg/mL范围 内与ΔA存在良好的线性关系,检出限0.2470μg/mL。
2)本方法测定受壳聚糖的分子量的影响,在实际样品测定中,需考虑采用 相近分子量的标准品做定量标准,具有试剂廉价,灵敏度高,重现性好和操作简 便等优点。
附图说明
图1 CTS-萘酚绿B-吐温20体系的吸收光谱图。
图2缓冲体系不同pH对于吸光度的影响图。
图3为醋酸-醋酸钠(NaAc-HAc)缓冲溶液的加入量对于吸光度值的影响。
图4为萘酚绿B溶液的加入量对于体系吸光度的影响。
图5不同增敏剂对于体系吸光度的影响。
图6吐温20的加入量对于体系吸光度的影响。
图7试剂加入顺序对于体系吸光度的影响。
图8反应温度对于体系吸光度的影响。
图9离子强度对于体系吸光度的影响
具体实施方式
以下通过具体实施例进一步描述本发明,但所述发明并不以任何方式限定本 发明专利保护的范围。
应用例
一种通过紫外分光光度法准确测定壳聚糖含量的方法,其包括下述步骤:
1)绘制ΔA和不同分子量的壳聚糖浓度的标准曲线
向10mL比色管中依次加入1.0mL一定浓度梯度的低分子壳聚糖标准溶液, 1.5mL浓度为2.0×10
2)10μg/mL的样品工作液的制备:将澳利维甲壳素胶囊去胶囊壳,称取0.04g 于100mL容量瓶中,用0.5mol/L冰醋酸溶解,定容得样品储备液。用漏斗脱脂 棉过滤储备液,滤液经6000r/min,离心机离心20min,取上清液2.5mL于100mL 容量瓶,定容,得浓度为10μg/mL的样品工作液。
3)根据壳聚糖分子量选择标准曲线:使用步骤1)种检测方法绘制ΔA和样 品工作液的标准曲线,并将其与不同分子量的壳聚糖标准曲线比较,确定相对应 的壳聚糖的分子量所使用的回归方程;
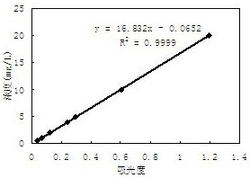
用样品工作液按实验方法绘制样品曲线,与不同分子量的壳聚糖标准曲线比 较,结果为样品的线性回归方程是ΔA=0.0222c-0.0001,相关系数0.9983,结 果与中分子量的壳聚糖标准曲线接近,采用中分子量壳聚糖的标准曲线作为定量 标准曲线。
按顺序依次加入1.00mL浓度为10μg/mL的样品工作液,1.50mL浓度为 2.0×10
表4样品分析结果
一种通过紫外分光光度法准确测定壳聚糖含量的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0