






专利摘要
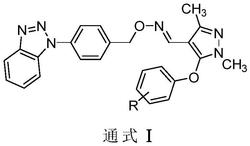
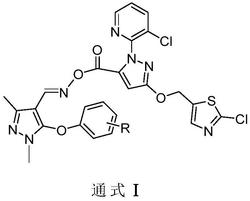
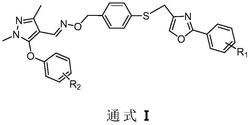
本发明公开了一种新型吡唑酰胺类化合物,结构如式Ⅰ所示:,式中各取代基团的定义见说明书。式Ⅰ化合物具有杀虫、杀菌作用,可用于防治农业虫害和病害。
权利要求
1.一种新型吡唑酰胺类化合物,结构式如式Ⅰ所示:
式中:
R1选自H或Cl;
R2选自Cl、Br、卤代乙氧基;
X1选自Cl或F;
X2选自H或Cl。
2.一种按照权利要求Ⅰ所述的通式Ⅰ化合物控制虫害或病害的用途。
3.一种杀虫、杀菌组合物,含有权利要求Ⅰ所述的通式Ⅰ化合物为活性组分和农业上可接受的载体。
说明书
技术领域本发明属于农用杀虫、杀菌剂领域,具体涉及一种新型吡唑酰胺类化合物及其用途。
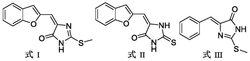
背景技术农业虫害或病害导致作物减产或品质降低。大量频繁地使用杀虫剂、杀菌剂易导致抗药性的产生。开发新的杀虫、杀菌剂品种是治理抗药性的有效途径。US5039693公开了下列化合物(KC)的制备及杀虫活性。该化合物商品名称为唑虫酰胺,对小菜蛾、蓟马及菜小绿叶蝉等害虫有很好的防治效果。
在现有技术中,如本发明所示的新型吡唑酰胺类化合物及其杀虫、杀菌活性未见公开。发明内容本发明的目的是提供一种结构新颖的吡唑酰胺类化合物,它可用于农业常见的虫害或病害的防治。
本发明的技术方案如下:
本发明提供了一种新型吡唑酰胺类化合物,结构通式如式Ⅰ所示:
式中:R1选自H或Cl;R2选自Cl、Br或卤代乙氧基;X1选自Cl或F;X2选自H或Cl。
本发明的通式Ⅰ化合物可由以下方法制备,反应式中各基团定义同前。
化合物Ⅱ与通式Ⅲ化合物在适宜的溶剂中,温度20~120℃下反应1~24h制得目标化合物Ⅰ。适宜的溶剂选自二氯甲烷、二氯乙烷、乙酸乙酯、乙腈、甲苯、N,N-二甲基甲酰胺、二甲基亚砜等。
反应过程中加入适量的有机碱如三乙胺、吡啶或N,N-二甲基甲酰胺等有利于缩短反应时间。
化合物Ⅱ和通式Ⅲ化合物的制备见本发明合成实例。
表1列出了部分通式Ⅰ化合物的结构和物理性质。
表1部分通式Ⅰ化合物的结构和物理性质
本发明的优点和积极效果:同已知的吡唑酰胺化合物(KC)相比,本发明的通式Ⅰ化合物的活性结构为吡啶环和吡唑环两类氮杂环的结合体,符合氮杂环类农药发展趋势要求,具有结构独特、新颖的特点。本发明化合物不仅杀虫活性高,出人意料的是本发明化合物同时具有很好的杀菌活性,在防治农业虫害的同时,对农作物常见真菌病害有兼治作用,有利于降低防治成本及节约劳动力成本;本发明化合物合成方法简单、安全,避免了唑虫酰胺(KC)合成过程需要高压加氢带来的操作安全性问题和生产成本高的弊端。因此,本发明化合物具有生产操作安全方便、生产成本低、综合防治效果好等优点,作为防治农业病虫害的农药新品种,在保障农业生产丰收及抗药性治理方面具有很好商品化价值和应用潜力。因此,本发明的通式Ⅰ化合物还包括控制病虫害的用途。
本发明的化合物在防治病虫害时,可根据实际需要单独使用,也可与其他活性物质组合使用,以提高产品的综合性能。
本发明还包括通式Ⅰ化合物作为活性组分的杀虫、杀菌组合物以及农业上可接受的载体。
本发明的组合物可以制剂的形式来施用:通式Ⅰ化合物作为活性组分溶解或分散于载体或溶剂中,添加适量的表面活性剂研制成乳油、悬浮剂或可湿性粉剂。
应明确的是,在本发明的权利要求所限定的范围内,可进行各种变换和改动。
具体实施方式
下列合成实例及生测试验结果可用来进一步说明本发明,但不意味着限制本发明。
合成实例
实例1、化合物Ia的制备
(1)3aH-吡唑并[3,4-b]吡啶-3-胺的合成
在250mL三口瓶中加入15.2g(0.11mol)2-氯-3-氰基吡啶和50mL乙醇,搅拌下缓慢滴入30mL80%水合肼,升温回流3h,降至室温,出现大量固体,抽滤,水洗,烘干,得到黄色片状固体13.8g,收率95%。
(2)1-(3-氯吡啶-2-基)-3-溴-4,5-二氢-1H-吡唑-5-甲酸乙酯的合成
向100mL的三口瓶中分别加入13.6g(0.05mol)1-(3-氯吡啶-2-基)-3-羟基-4,5-二氢-1H-吡唑-5-甲酸乙酯和50mL乙腈,滴入10.0g(0.35mol)三溴氧磷,回流3-4h,薄层板(TLC)检测原料点消失,减压蒸馏除去大部分乙腈,加入饱和碳酸氢钠水溶液直到没有气泡产生,至pH值为8-9,搅拌0.5h,产生大量颗粒状固体,抽滤,水洗,烘干称重13.9g,收率84.1%。以甲醇在-20℃重结晶,得到青绿色晶体。
(3)1-(3-氯吡啶-2-基)-3-溴-1H-吡唑-5-甲酸乙酯的合成
向100mL三口瓶中加入13.3g(0.04mol)1-(3-氯吡啶-2-基)-3-溴-4,5-二氢-1H-吡唑-5-甲酸乙酯和乙腈40mL,缓慢滴入4mL浓硫酸,搅拌10min,分次加入16.2g(0.06mol)过硫酸钾,升温至回流,反应2h,降温,蒸出大部分溶剂,加入20mL水。搅拌出现黄色固体,抽滤,干燥,称重11.8g,收率89%。
(4)1-(3-氯吡啶-2-基)-3-溴-1H-吡唑-5-甲酸的合成
向100mL反应瓶中分别加入12g(0.03mol)1-(3-氯吡啶-2-基)-3-溴-1H-吡唑-5-甲酸乙酯和30mLg甲醇,搅拌下滴入20%的NaOH溶液14mL,在40~50℃下反应2h,蒸出甲醇,降温,滴入浓盐酸调pH=2~3,析出白色固体,水洗,抽滤,真空干燥,得白色固体11g,收率90%。
(5)1-(3-氯吡啶-2-基)-3-溴-1H-吡唑-5-甲酰氯的合成
向100mL反应瓶中加入9.1g(0.03mol)1-(3-氯吡啶-2-基)-3-溴-1H-吡唑-5-甲酸和30mL甲苯,滴入10mL氯化亚砜,回流反应4h,蒸出溶剂,得到橙红色液体9.0g,收率95%
(6)1-(3-氯吡啶-2-基)-3-溴-N-(4aH-吡咯并[3,4-b]吡啶-5-基)-1H-吡唑-5-甲酰胺的合成
向100mL三口瓶中加入2.1g(0.016mol)3-aH-吡唑并[3,4-b]吡啶-3-胺和15mL乙腈,常温搅拌下缓慢滴入20mL乙腈和5.6g(0.017mol)1-(3-氯吡啶-2-基)-3-溴-1H-吡唑-5-甲酰氯的混合液,出现大量红色固体,反应1.5h后,抽滤,用3×5mL乙腈洗涤,干燥得到5.3g红色固体,收率80%。以甲醇重结晶得到红色晶体,m.p.:231-234℃。1HNMR(500MHz,DMSO-d6):6.30(1H,s,-CH),6.92-6.93(1H,d,-CH),7.56-7.57(6H,m,-ArH),8.51(1H,s,-NH)。
实例2、化合物If的制备
(1)1-(3-氯吡啶-2-基)-3-溴-4-氯-1H-吡唑-5-甲酸乙酯的合成
向100mL反应瓶中加入11.0g(0.03mol)1-(3,5-二氯吡啶-2-基)-3-溴-4,5-二氢-1H-吡唑-5-甲酸乙酯和30mL乙腈,搅拌下滴入9.2g(0.07mol)磺酰氯,常温反应2h,薄层板(TLC)检测原料消失,蒸出溶剂,得黄色油状液体10g,收率82%。直接用于下一步反应。
(2)1-(3,5-二氯吡啶-2-基)-3-溴-4-氯-1H-吡唑-5-甲酸的合成
向100mL反应瓶中分别加入0.03mol(11g)1-(3,5-二氯吡啶-2-基)-3-溴-4-氯-1H-吡唑-5-甲酸乙酯和30mL甲醇,搅拌下滴入20%的NaOH溶液14mL,在40~50℃下反应2h,蒸出甲醇,降温,滴入浓盐酸调pH=2~3,析出白色固体,水洗,抽滤,真空干燥,得白色固体11g,收率91%。
(3)1-(3,5-二氯吡啶-2-基)-3-溴-4-氯-1H-吡唑-5-甲酰氯的合成
向100mL反应瓶中加入10.1g(0.03mol)1-(3,5-二氯吡啶-2-基)-3-溴-4-氯-1H-吡唑-5-甲酸和30mL甲苯,滴入10mL氯化亚砜,回流反应4h,蒸出溶剂,得到橙红色液体10.1g,收率95%
(4)1-(3,5-二氯吡啶-2-基)-3-溴-4-氯-N-4aH-吡咯并[3,4-b]吡啶-5-基)-1H-吡唑-5-甲酰胺(If)的合成
向100mL三口瓶中加入2.1g(0.016mol)3-aH-吡唑并[3,4-b]吡啶-3-胺和10mL乙腈,常温搅拌下缓慢滴入20mL乙腈和6.1g(0.017mol)1-(3,5-二氯吡啶-2-基)-3-溴-4-氯-1H-吡唑-5-甲酰氯的混合液,出现大量红色固体,反应1.5h后,抽滤,用3×5mL乙腈洗涤,干燥得到5.4g红色固体,收率75%。以乙酸乙酯和石油醚重结晶得到红色晶体,m.p.223-227℃。
1HNMR(500MHz,DMSO-d6):6.43(1H,s,-CH),7.45-8.33(5H,m,-ArH),8.66(1H,s,-NH)。
实例3、化合物Ig的制备
(1)1-(3-氯吡啶-2-基)-3-羟基-1H-吡唑-5-甲酸乙酯的合成
向100mL三口瓶中加入10.8g(0.04mol)2-(3-氯吡啶-2-基)-5-羟基-1,4-二氢-2H-吡唑-3-羧酸乙酯和乙腈30mL,慢慢滴入4mL浓硫酸,搅拌10min,加入过硫酸钾16.2g(0.06mol),升温至回流,反应2h,降温,蒸出大部分溶剂,加入20mL水。搅拌出现黄色固体,抽滤,干燥,称重9.74g,收率91%。
(2)1-(3-氯吡啶-2-基)-3-(2-溴乙氧基)-1H-吡唑-5-甲酸乙酯的合成
向100mL的三口瓶中分别加入13.4g(0.05mol)1-(3-氯吡啶-2-基)-3-羟基-1H-吡唑-5-甲酸乙酯,8.95g(0.07mol)碳酸钾和50mL乙腈,滴入13.2g(0.07mol)1,2-二溴乙烷,回流7-8h,薄层板检测原料点消失,抽滤,以3×10mL乙腈洗涤滤渣,收集滤液,蒸出溶剂得红色油状物,直接用于下一步反应。
(3)1-(3-氯吡啶-2-基)-3-(2-溴乙氧基)-1H-吡唑-5-甲酸的合成
向100mL反应瓶中分别加入0.03mol(11.2g)1-(3-氯吡啶-2-基)-3-羟基-1H-吡唑-5-甲酸乙酯和40mL甲醇,搅拌下滴入20%的NaOH溶液15mL,在40~50℃下反应2h,蒸出甲醇,降温,滴入浓盐酸调pH=2~3,析出白色固体,水洗,抽滤,真空干燥,得白色固体9.5g,收率91%。
(4)1-(3-氯吡啶-2-基)-3-(2-溴乙氧基)-1H-吡唑-5-甲酰氯的合成
向100mL反应瓶中加入10.4g(0.03mol)1-(3-氯吡啶-2-基)-3-羟基-1H-吡唑-5-甲酸和30mL甲苯,滴入10mL氯化亚砜,回流反应4h,蒸出溶剂,得到橙红色液体10.4g,收率95%。
(5)1-(3-氯吡啶-2-基)-3-(2-溴乙氧基)-N-(4aH-吡咯并[3,4-b]吡啶-5-基)-1H-吡唑-5-甲酰胺(Ig)的合成
向100mL三口瓶中加入2.1g(0.016mol)3aH-吡唑并[3,4-b]吡啶-3-胺和10mL乙腈,常温搅拌下缓慢滴入20mL乙腈和6.2g(0.017mol)1-(3-氯吡啶-2-基)-3-羟基-1H-吡唑-5-甲酰氯的混合液,出现大量红色固体,反应1.5h后,抽滤,用3×5mL乙腈洗涤,干燥得到5.5g红色固体,收率75%。以乙酸乙酯重结晶得到红色晶体,m.p.235-238℃。1HNMR(500MHz,DMSO-d6):3.91-3.92(2H,t,-CH2),4.51-4.52(2H,t,-CH2),7.66-8.32(8H,m,-PyH),8.74(1H,s,-NH)。
按照上述实例1、实例2及实例3的方法,可以制备表1中其他通式Ⅰ化合物。1HNMR(500MHz,DMSO-d6),δ(ppm)数据如下:
化合物Ib:4.13-4.14(2H,t,-CH2),4.51-4.52(2H,t,-CH2),4.93(1H,s,-CH),6.65-6.66(1H,d,-CH),7.69-8.43(5H,m,-PyH),8.67(1H,s,-NH);
化合物Ic:6.01(1H,s,-CH),6.56-6.57(1H,d,-CH),7.45-8.15(6H,m,-PyH),8.62(1H,s,-NH);
化合物Id:5.76(1H,s,-CH),6.45-6.46(1H,d,-CH),7.76-8.45(5H,m,-PyH),8.49(1H,s,-NH);
化合物Ie:5.86(1H,s,-CH),6.33-6.34(1H,d,-CH),7.32-8.01(5H,m,-PyH),8.95(1H,s,-NH);
化合物Ih:3.76-3.77(2H,t,-CH2),4.57-4.59(2H,t,-CH2),5.44-5.45(1H,d,-CH),6.99(1H,s,-CH),7.36-8.11(5H,m,-PyH),8.43(1H,s,-NH);
化合物Ii:3.92-3.93(2H,t,-CH2),4.95-4.96(2H,t,-CH2),6.13-6.15(1H,d,-CH),6.57(1H,s,-CH),7.29-8.25(6H,m,-PyH),8.65(1H,s,-NH);
化合物Ij:3.85-3.87(2H,t,-CH2),4.69-4.70(2H,t,-CH2),5.93-5.94(1H,d,-CH),6.22(1H,d,-CH),7.56-8.37(5H,m,-PyH),8.79(1H,s,-NH)。
生测实例
实例4、甜菜夜蛾生物活性测试
杀甜菜夜蛾活性测试方法:本发明化合物对甜菜夜蛾活性测定采用国际抗性行动委员会(IRAC)提出的浸叶法。用配制好的待测药液,用直头眼科镊子浸渍甘蓝叶片,时间3-5秒,甩掉余液,每次1片,每个样品共3片,按样品标记顺序依次放在处理纸上。待药液干后,放入具有标记的10cm长的直型管内,接入2龄甜菜夜蛾幼虫30头,用纱布盖好管口。将试验处理置于标准处理室内,48h检查结果以拔针轻触虫体,不动者为死亡。计算死亡率。(试验做3次重复,取平均值)
在供试化合物中,表1中化合物在浓度10ppm时对甜菜夜蛾有较高防效,死亡率达90%。
部分供试化合物中,下列化合物在浓度0.1ppm时,对甜菜夜蛾有较高防效,死亡率达90%以上:Ia,Id,If,Ig。
按照以上方法,选取化合物Ⅰa和已知化合物KC进行杀甜菜夜蛾活性平行比较,实验结果见表2。
表2化合物1与已知化合物KC杀甜菜夜蛾活性数据
实例5、离体杀菌普筛
方法:采用活体盆栽测定方法。待测化合物原药用少量丙酮溶解,用含有0.1%吐温-80的水稀释至所需的浓度。喷雾施药到植物试材上,24小时后进行病害接种。接种后,将植物放在恒温恒湿培养箱中,使感染继续,待对照充分发病后(通常为一周时间),进行评估调查。
部分测试结果如下:药液浓度为15ppm时,化合物Ia,Ic,Ie,If等对水稻稻瘟病的防治效果达到60%以后,药液浓度为300ppm时,化合物表Ia,Ih等对黄瓜霜霉病防治效果达到95%,对炭疽病防治效果达到92%。
一种吡唑酰胺类化合物及其用途专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。













动态评分
0.0