

IPC分类号 : C07D285/06,C07D417/12,C07D473/00,C07D417/14,A01N47/34,A01N47/36,A01P7/04,A01P7/02,A01P3/00,A01P1/00







专利摘要
本发明提供了一类含1,2,3-噻二唑基团的甲酰脲衍生物及其制备方法和用途,本发明涉及含1,2,3-噻二唑的杂环化合物,它们具有如下的化学结构通式: 本发明公开了上述化合物的结构通式、合成方法与用作杀虫剂、杀菌剂、抗植物病毒剂、植物激活剂的用途,其与农业上可接受的助剂或增效剂混合用于制备杀虫剂、杀菌剂、抗植物病毒剂、植物激活剂的工艺;还公开了这些化合物与商品杀虫剂、杀菌剂、抗植物病毒剂、植物激活剂组合使用在防治农业、林业、园艺病害、虫害、病毒病害中的用途和制备方法。
说明书
技术领域技术领域
本发明的技术方案涉及含1,2-二唑化合物,具体涉及4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物。
技术背景背景技术
苯甲酰基脲类化合物具有广泛的生物活性,3-溴代丙酰胺苯甲酰脲具有医药的生物活性(李建农,等.药学学报.2004,39(7):491-495;Song Dan-Qing,et al.Bioorg.Med.Chem.Let.,2009,19:755-758);在农药方面,自1972年发现苯甲酰基脲(BPUs)类化合物对昆虫的蜕皮具有抑制作用来,已有10多个商品化品种面世,BPUs类杀虫剂的研究曾获得2000年度美国“总统绿色化学挑战奖”(闫蒙钢,等.化学教育,2009,5:75-77);BPUs类昆虫生长调节剂属几丁质合成抑制剂,干扰靶标昆虫几丁质合成而导致其死亡,或直接降解昆虫几丁质;在甲酰基N-末端进行适当的芳基取代,不仅能增强它的杀虫活性,还能改变幼虫有丝分裂阻滞带的成分,影响靶标昆虫孵育。该类杀虫剂对未成熟阶段害虫活性高,被广泛用于柑橘、棉花、葡萄、大豆、果树、玉米和咖啡上,防治植食性害虫。由于这类杀虫剂的水溶性极低,因此持效作用好(米娜,等.世界农药,2009,31(增刊II):24-26)。BPUs类化合物具有抗蜕皮激素的生物活性,能增加几丁质酶的催化活性,加速几丁质的酶解过程,还可抑制几丁质合成酶(CS)的活性。对于幼虫,若CS活性被抑制,不仅抑制了几丁质链的延长,还削弱了对O-糖苷键的保护作用,干扰蛋白质和几丁质的结合,严重破坏昆虫蜕皮,阻止围食膜的形成,影响正常发育,结果使表皮松软缺乏硬度,最终导致昆虫脱水死亡或成畸形蛹死亡,昆虫的表皮是其“骨骼”,也是昆虫气体交换的重要组织,没有“骨骼”且气体交换功能失常的昆虫就同身患软骨病和呼吸障碍的病人。对于卵,若CS活性受抑制,则影响卵的呼吸代谢及胚胎发育中DNA和蛋白质代谢,使卵内幼虫缺乏几丁质而不能孵化或孵化后死亡,所以,它也是不育剂(Cohen et al.Annu Rev Entomo,1987,2(3):71-93;Merzendorfer.Comp.Physiol.B.2006.176:1-15;梁英,等.农药,2009,48(9):625-632;Matsumura et al.Pestic.Biochem.Physiol.,doi:10.1016/j.pestbp.2009.101)。BPUs类杀虫剂的先导结构见式1:
式1BPUs类杀虫剂的结构通式
商品化品种的创制分析如下:在苯甲酰基上的修饰引入卤素获得杀虫活性很好的除虫脲、灭幼脲等品种;在芳胺环上引入CF3、OCF3、OCF2CF2等得到除虫隆、氟铃脲等品种;在芳胺环上引入多个卤原子开发出伏虫隆;在芳胺环对位引入取代芳(杂)氧基开发出啶虫隆、氟虫脲等品种。
BPUs类几丁质合成抑制剂的主要局限在于:其杀虫谱窄,不能同时兼治同一种作物上的多种害虫;杀虫速度慢不能迅速控制害虫,昆虫中毒后仍能取食直到下次脱皮,对鳞翅目昆虫的致死时间需2-7d,速效性差;对水生甲壳类生物的生长不利。其创制的主要方向是引入卤素以提高BPUs类几丁质合成抑制剂的触杀活性;引入强极性基团以提高内吸或内渗杀虫活性。系统的结构衍生主要在两个芳香环和脲桥上进行。在苯甲酰脲类化合物中引入硫肟醚合成的化合物具有生物活性(陈灿,等.河北化工.2005,3:31-32)。最近的研究发现,具有邻氨基苯甲酸结构的苯甲酰脲类化合物对甜菜夜蛾表现较好的防治效果(彭永武,等.农药,2009,48(5):326-328)。含杂环如1,3,4-噻二唑结构的苯甲酰脲类化合物对淡色库蚊幼虫具有较好的杀虫活性(李兴海,等.化学通报,2003,5:333-336),而1,3,4-噁二唑的苯甲酰脲类化合物由于溶解性没有得到解决而影响了其生物活性(张文文,等.农药学学报,2009,11(1):36-40);在分子中引入吡嗪后对蚊幼虫显示较好的活性(Sun Ranfeng,et al.J.Agric.Food Chem.,2009,57(14):6356-6361)。卤素尤其是F原子引入后的苯甲酰脲类化合物对鳞翅目的棉花害虫和蔬菜害虫显示出很好的生物活性(Zhang Jian,et al.J.Agric.FoodChem.,2009,58(5):2736-2740)。
1,2,3-噻二唑衍生物有广泛的生物活性,含1,2,3-噻二唑活性结构单元的专利和文献均有较为详细的总结(Bakulev,et al.Newyork:John Wiley & Sons,Inc,2004)。1,2,3-噻二唑衍生物在医药和农药中商品化的品种不多,农业领域应用的只有棉花脱叶剂---脱叶灵(N-苯基-N’-1,2,3-噻二唑-5-脲,TDZ)、植物激活剂---活化酯(苯并-1,2,3-噻二唑-7-硫代羧酸甲酯,BTH)、稻田杀菌剂---噻酰菌胺(3’-氯-4,4’-二甲基-1,2,3-噻二唑-5-甲酰苯胺,TDL)。含1,2,3-噻二唑的甲酰脲类衍生物及其生物活性未见报道。
为了寻找更高生物活性的1,2,3-噻二唑类新化合物,本发明设计合成了一类4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物,同时进行了系统的生物活性的筛选和评价,以期为新农药的创制研究提供侯选化合物。
发明内容发明内容
本发明所要解决的技术问题是:提供新的4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物的合成方法,提供这类化合物抑制农业和园艺以及林业植物病原物的生物活性及其测定方法,同时提供这些化合物在农业领域和园艺领域以及林业领域的中应用。
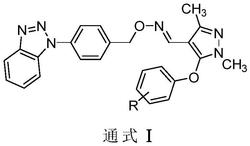
本发明解决该技术问题所采用的技术方案是:具有农业、园艺和林业杀菌活性的4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物的化学结构通式见式V:
其中,R为选自对硝基苯基、邻硝基苯基、2,4-二硝基苯基、4-甲基嘧啶-2-基、吡啶基-3-甲基、2,4,5-三氯苯基、2-萘基、2,5-二氯苯基、吡啶-2-基、吡啶-3-基、环己基、正辛基、2-氨基苯基、3-氟-4-甲基苯基、正丙基、苯并咪唑-2-基、3,5-二氯苯基、4,6-二甲氧基 嘧啶-2-基、对溴苯基、4-氯-6-甲氧基嘧啶-2-基、4,6-二甲基嘧啶-2-基、4-乙氧羰基-1,2,3-噻二唑-5-基、4,6-二氯嘧啶-2-基、2,6-二氯嘧啶-4-基、2-甲基-4-氯苯基、2,5-二甲氧基嘧啶-4-基、3-氯苯基硫代甲酰胺基、7H-嘌呤-2-基、5-氯吡啶-2-基、4-甲基-6-溴吡啶-2-基、4-甲基吡啶-2-基、环丙基甲基、环丙基、环丁基、2-三氟甲基-5-氯苯基、2-氯-4-三氟甲基苯基、6-甲氧基苯并[d]噻唑-2-基、6-甲基苯并[d]噻唑-2-基、4-甲基苯并[d]噻唑-2-基、6-硝基苯并[d]噻唑-2-基、2-甲基苯并[d]噻唑-6-基、6-溴苯并[d]噻唑-2-基、2-氯苯基、4-氯苯基、3-硝基苯基、4-乙基苯基、3-氯苯基、3-氯-4-甲基苯基、3,5-二氯-4-(1,1,2-三氟乙氧基)苯基、3-胺基苯基、3-甲基吡啶-2-基、2-氯吡啶-3-基、6-甲基吡啶-2-基、叔丁基、嘧啶-2-基、5-甲基-1,3-噻唑-2-基、5-硝基-1,3-噻唑-2-基、1,3,4-噻唑-2-基、1,3-噻唑-2-基、6-甲氧羰基吡嗪-2-基、5-溴吡嗪-2-基、5-氯吡嗪-2-基、3-甲氧基-5-溴吡嗪-2-基、吡嗪-2-基、3,5-二溴吡嗪-2-基、3,5-二溴-4-甲基吡啶-2-基、4-三氟甲氧基苯基、3,5-二溴吡啶-4-基、5-溴吡啶-2-基、5-溴-1,3,4-噻二唑-2-基、3-溴吡啶-4-基、6-氯-2,1,3-苯并噻二唑-5-基、3-苯基-1,2,4-噻二唑-5-基、5-(4-甲基-1,2,3-噻二唑-5-基)-1,3,4-噻二唑-2-基、2,4-二甲基苯基的基团。
本发明的4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物Ⅴ的合成方法如下:
其中:R为选自如上所述的任何一种基团。
具体分为以下步骤:
A.中间体4-甲基-1,2,3-噻二唑-5-甲酰氯Ⅰ的制备:
4-甲基-1,2,3-噻二唑-5-甲酰氯Ⅰ的制备参照中国专利200610013185.5的方法进行。
将0.067摩尔的4-甲基-1,2,3-噻二唑-5-甲酸和29毫升二氯亚砜加入到100毫升三口圆底烧瓶中,80摄氏度下加热回流6小时,减压蒸除过量的二氯亚砜,减压蒸馏在2000Pa下收集94-96摄氏度的馏分得淡黄色产物9.25克,收率85%,中间体4-甲基-1,2,3-噻二唑-5-甲酰氯Ⅰ密封保存在干燥器中备用,4-甲基-1,2,3-噻二唑-5-甲酰氯I制备的量按相应比例扩大或缩小。
B.4-甲基-1,2,3-噻二唑-5-甲酰胺Ⅱ的制备:
在500毫升的两口瓶中,加入30毫升25%的氨水,5毫升的三乙胺,50毫升的四氢呋喃,在冰浴的冷却并机械搅拌下滴加5克经10毫升四氢呋喃稀释过的4-甲基-1,2,3-噻二唑-5-甲酰氯Ⅰ,15分钟滴加完毕,冰浴下继续搅拌2小时,然后在室温下搅拌7小时。反应完毕后,用分液漏斗分液,15毫升四氢呋喃萃取水层3次,合并有机层,用无水硫酸钠干燥过夜,抽滤除去无水硫酸钠,旋转蒸发除去溶剂,有白色片状固体生成,为4-甲基-1,2,3-噻二唑-5-甲酰胺Ⅱ;合成化合物Ⅱ的量按相应比例扩大或缩小。
C.4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯Ⅲ的制备:
取100毫升的三口瓶,加入2克4-甲基-1,2,3-噻二唑-5-甲酰胺Ⅱ以及15毫升的1,2-二氯乙烷,搅拌,使酰胺在溶剂中分散均匀,在冰浴下用滴液漏斗滴加4.3克经5毫升1,2-二氯乙烷稀释过的草酰氯,30分钟滴加完毕,然后室温下搅拌1小时,再加热至80摄氏度回流7小时,反应液从白色变浑浊转变为浅黄色透明溶液,加热后颜色逐渐变深。反应完毕后,旋转蒸发除去溶剂得产物4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯Ⅲ,无须进一步纯化直接用于后续反应。合成化合物Ⅲ的量按相应比例扩大或缩小。
D.4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物Ⅴ的制备:
在100毫升圆底烧瓶中加入胺类化合物RNH2IV,再滴加1,2-二氯乙烷并搅拌至胺类化合物刚好完全溶解为止,然后在搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯Ⅲ(约0.5克)(按摩尔比胺∶异氰酸酯为0.9∶1),15分钟滴加完毕,然后加热回流1小时,在室温下搅拌8小时后反应完毕,静置反应混合物,抽滤分离固体与溶剂,收集固体即为粗产物V;滤液放入冰箱中冷却后有固体析出,合并固体并用洗涤干燥,用体积比为1∶3的乙酸乙酯∶石油醚洗涤得产物纯品;用所得纯品计算收率,测定熔点和1H NMR的测定,合成化合物Ⅴ的量按相应比例扩大或缩小;化合物化学结构见表1。
E.4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物Ⅴ对病原真菌生长活性影响的测定:
本发明的4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物Ⅴ杀菌或抑菌活性采用菌体生长率测定法,具体过程是,取5毫克样品溶解在适量二甲基甲酰胺内,然后用含有一定量吐温20乳化剂水溶液稀释至500微克/毫升的药剂,将供试药剂在无菌条件下各吸取1毫升注入培养皿内,再分别加入9毫升培养基,摇匀后制成50微克/毫升含药平板,以添加1毫升灭菌水的平板做空白对照,用直径4毫米的打孔器沿菌丝外缘切取菌盘,移至含药平板上,呈等边三角形摆放,每处理重复3次,将培养皿放在24±1摄氏度恒温培养箱内培养,待对照菌落直径扩展到2-3厘米后调查各处理菌盘扩展直径,求平均值,与空白对照比较计算相对抑菌率,供试菌种包括多种农业上常见植物病原菌,如:F:番茄早疫病菌(Alternariasolani);D:花生褐斑病菌(Cercospora arachidicola);I:苹果轮纹病菌(Physalospora piricola);L:黄瓜灰霉病菌(Botrytis cinerea);T:水稻纹枯病菌(Pellicularia sasakii);G:小麦赤霉病菌(Gibberella zeae);AK:马铃薯晚疫病菌(Phytophthora infestans(Mont.)de Bary);N:油菜菌核病菌(Sclerotinia sclerotiorum);RC:禾谷丝核菌(Rhizoctonia cerealis)。
F.本发明的4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物Ⅴ对小菜蛾杀虫活性的测定:
本发明的4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物Ⅴ对小菜蛾的杀虫活性的筛选方法如下:采用叶片药膜法;样品先用200毫克/升进行活性初筛。原药先用少量丙酮溶解,然后用0.5‰Triton-100稀释,0.5‰Triton-100水为对照,每个浓度4次重复,每个重复处理10头左右试虫;取新鲜无污染的甘蓝叶片,在系列浓度梯度的药液中浸10秒,于室内晾干(约2小时)后,放入直径9厘米的培养皿中,分别接入大小基本一致的小菜蛾2龄初期幼虫;用橡皮筋扎紧后置于小菜蛾恒温养虫室中,96小时或120小时后检查结果并计算校正死亡率;以小毛笔或镊子轻触虫体,不能协调运动为死亡;以灭幼脲为阳性对照。
G.本发明的4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物Ⅴ对蚊幼虫杀虫活性的测定:
本发明的4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物Ⅴ对蚊幼虫的杀虫活性的筛选方法如下:尖音库蚊淡色亚种(Culex pipiens pallens),室内饲养的正常群体;称取供试化合物2.5毫克于盘尼西林药瓶中,加10毫升丙酮,振荡溶解样本配制成250微克/毫升的母液;移取1毫升母液于盛有49毫升水的100毫升烧杯中,选取10头4龄初蚊幼虫,连同10毫升饲养液一并倒入烧杯中,其药液浓度为5微克/毫升。将处理好的溶液连同蚊幼虫的烧杯放入标准处理室内保持温度为25摄氏度培养,24小时后开始检查结果,每天加少量蚊饲料并吸出蚊蛹,补充烧杯中蒸发的水分(5毫升/天),直到蚊幼虫全部死亡或化蛹;多数幼虫在8天内化蛹;以含1毫升丙酮的水溶液为空白对照;以灭幼脲为阳性对照。
本发明的有益效果是:本发明对4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物进行了先导结构的优化,并对合成的新化合物进行了抑菌活性和杀虫活性的筛选,这类化合物可以用于防治农业领域和林业领域以及园艺领域的植物病害或植物虫害的防治。
本发明将通过特定制备和生物活性测定实施例更加具体地说明4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物Ⅴ的合成和生物活性及其应用,但所述实施例仅用于具体的说明本发明而非限制本发明,尤其是其生物活性仅仅是举例说明,而非限制本专利,具体实施方式如下:
附图说明具体实施方式实施例1
中间体4-甲基-1,2,3-噻二唑-5-甲酰胺Ⅱ的制备
在500毫升的两口瓶中,加入30毫升25%的氨水,5毫升的三乙胺,50毫升的四氢呋喃,在冰浴的冷却并机械搅拌下滴加5克经10毫升四氢呋喃稀释过的4-甲基-1,2,3-噻二唑-5-甲酰氯Ⅰ,15分钟滴加完毕,冰浴下继续搅拌2小时,然后在室温下搅拌7小时。反应完毕后,用分液漏斗分液,15毫升四氢呋喃萃取水层3次,合并有机层,用无水硫酸钠干燥过夜,抽滤除去无水硫酸钠,旋转蒸发除去溶剂,有白色片状固体生成,为4-甲基-1,2,3-噻二唑-5-甲酰胺Ⅱ。
实施例2
中间体4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯Ⅲ的制备
取100毫升的三口瓶,加入2克4-甲基-1,2,3-噻二唑-5-甲酰胺Ⅱ以及15毫升的1,2-二氯乙烷,搅拌,使酰胺在溶剂中分散均匀,在冰浴下用滴液漏斗滴加4.3克经5毫升1,2-二氯乙烷稀释过的草酰氯,30分钟滴加完毕,然后室温下搅拌1小时,再加热至80摄氏度回流7小时,反应液从白色变浑浊转变为浅黄色透明溶液,加热后颜色逐渐变深。反应完毕后,旋转蒸发除去溶剂得产物4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯Ⅲ,无须进一步纯化直接用于后续反应。
实施例3
化合物GDD-1:N-(4-硝基苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔对硝基苯胺和20毫升1,2-二氯乙烷,搅拌至对硝基苯胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离得固体产 物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:178-180摄氏度,收率58%。核磁数据(DMSO-d6,化学位移):2.89(s,3H,CH3),7.91(s,1H,Ar-H),7.94(s,1H,Ar-H),8.31(s,1H,Ar-H),8.34(s,1H,Ar-H),10.71(s,1H,NH),11.68(s,1H,NH)。
实施例4
化合物GDD-2:N-(2-硝基苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔邻硝基苯胺和20毫升1,2-二氯乙烷,搅拌至邻硝基苯胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,抽滤分离固体与溶剂得固体,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱,析出固体,合并固体,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:158-160摄氏度,收率39%;核磁数据(DMSO-d6,化学位移):2.84(s,3H,CH3),7.41(t,1H,J=15.6Hz,CH),7.80(t,1H,J=15.6Hz,CH),8.18(d,1H,J=8Hz,CH),8.44(d,1H,J=8.4Hz,CH),11.77(s,1H,NH),11.84(s,1H,NH)。
实施例5
化合物GDD-3:N-(2,4-二硝基苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2,4-二硝基苯胺和20毫升1,2-二氯乙烷,搅拌至2-氨基-4-甲基嘧啶溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤得固体,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:164-166摄氏度,收率81%;核磁数据(DMSO-d6,化学位移):2.84(s,3H,CH3),8.61(d,J=9.2Hz,1H,Ar-H),8.79(d,J=9.2Hz,1H,Ar-H),8.897(s,1H,Ar-H),12.02(s,1H,NH),12.30(s,1H,NH)。
实施例6
化合物GDD-4:N-(4-甲基嘧啶-2-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氨基-4-甲基嘧啶和20毫升1,2-二氯乙烷,搅拌至2-氨基-4-甲基嘧啶溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤得固体,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点98-102摄氏度,收率57%;核磁数据(DMSO-d6,化学位移):2.24(s,3H,Ar-CH3),2.79(s,3H,CH3),6.48(d,1H,J=5.08Hz,Ar-H),8.03(s,1H,NH),8.09(d,1H,J=5.08Hz,Ar-H),8.24(s,1H,NH)。
实施例7
化合物GDD-5:N-(吡啶-3-甲基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔3-氨甲基吡啶和20毫升1,2-二氯乙烷,搅拌至3-氨甲基吡啶溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完后,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:113-116摄氏度,收率63%;核磁数据(DMSO-d6,化学位移):2.78(s,3H,CH3),4.45(d,2H,J=6.093Hz,CH2),7.38(m,1H,CH),7.75-7.69(m,1H,CH),8.46-8.48(m,1H,CH),8.56(s,1H,CH),8.77(t,1H,J=11.789Hz,NH),11.23(s,1H,NH)。
实施例8
化合物GDD-6:N-(2,4,5-三氯苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2,4,5-三氯苯胺和20毫升1,2-二氯乙烷,搅拌至2,4,5-三氯苯胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:122-124摄氏度,收率23%;核磁数据(DMSO-d6,化学位移):2.88(s,3H,CH3),8.05(s,1H,CH),8.59(s,1H,CH),11.03(s,1H,NH),11.98(s,1H,NH)。
实施例9
化合物GDD-7:N-(2-萘基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-萘胺和20毫升1,2-二氯乙烷,搅拌至2-萘胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:190-193摄氏度,收率65%;核磁数据(DMSO-d6,化学位移):2.90(s,3H,CH3),7.42-7.62(m,3H,CH),7.92-8.04(m,4H,CH),10.54(s,1H,NH),11.60(s,1H,NH)。
实施例10
化合物GDD-8:N-(2,5-二氯苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2,5-二氯苯胺和20毫升1,2-二氯乙烷,搅拌至2,5-二氯苯胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤得固体,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:186-190摄氏度,收率26%;核磁数据(DMSO-d6,化学位移):2.82(s,3H,CH3),7.24-7.25(m,1H,CH),7.62(d,1H,J=8.622Hz,CH),8.37(d,1H,J=2.426Hz,CH),10.97(s,1H,NH),11.86(s,1H,NH)。
实施例11
化合物GDD-9:N-(吡啶-2-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氨基吡啶和20毫升1,2-二氯乙烷,搅拌至2-氨基吡啶溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:182-184摄氏度,收率41%;核磁数据(DMSO-d6,化学位移):2.82(s,3H,CH3),7.16-7.19(m,1H,CH),7.83-7.88(m,1H,CH),7.97(d,1H,J=8.109Hz,CH),8.35(d1H,J=4.076Hz,CH),10.67(s,1H,NH),11.69(s,1H,NH)。
实施例12
化合物GDD-10:N-(吡啶-3-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔3-氨基吡啶和20毫升1,2-二氯乙烷,搅拌至3-氨基吡啶溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤得固体,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:173-176摄氏度,收率70%;核磁数据(DMSO-d6,化学位移):22.83(s,3H,CH3),7.39-7.42(m,1H,CH),8.03-8.05(m,1H,CH),8.33-8.34(m,1H,CH),8.75(d,1H,2.481Hz,CH),10.33(s,1H,NH),11.55(s,1H,NH)。
实施例13
化合物GDD-11:N-环己基-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔环己胺和20毫升1,2-二氯乙烷,搅拌至环己胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,没有沉淀产生,在室温下搅拌4小时后再加热到50摄氏度条件下回流4小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:179-181摄氏度,收率39%;核磁数据(DMSO-d6,化学位移):1.18-1.35(m,7H,CH2),1.53-1.56(m,1H,CH2),1.83-1.85(m,2H,CH2),2.83(s,3H,CH3),3.61(s,1HCH),8.13(d,1H,J=7.750Hz,NH),11.08(s,1H,NH)。
实施例14
化合物GDD-12:N-正辛基-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔正辛胺和20毫升1,2-二氯乙烷,搅拌至正辛胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,没有沉淀产生,在室温下搅拌4小时后再加热到80摄氏度条件下回流4小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点85-88摄氏度,收率47%;核磁数据(DMSO-d6, 化学位移):0.85-0.88(t,3H,CH3),1.24-1.28(m,10H,CH2),1.48-1.51(t,2H,CH2),2.77(s,3H,CH3),3.19-3.21(m,2H,CH2),821(s,1H,NH),11.11(s,1H,NH)。
实施例15
化合物GDD-13:N-(2-氨基苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔邻苯二胺和20毫升1,2-二氯乙烷,搅拌至苯二胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤得固体,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;收率86%,熔点:135-138摄氏度,核磁数据(DMSO-d6,化学位移):2.78(s,3H,CH3),6.81-6.86(m,1H,CH),6.96-6.98(m,1H,CH),7.25-7.28(m,1H,CH),7.71-7.74(m,1H,CH),10.22(s,1H,NH),11.64(s,1H,NH)。
实施例16
化合物GDD-14:N-(3-氟-4-甲基苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔3-氟-4-甲基苯胺和20毫升1,2-二氯乙烷,搅拌至3-氟-4-甲基苯胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:191-195摄氏度,收率85%;核磁数据(DMSO-d6,化学位移):2.82(s,3H,CH3),7.23-7.26(m,2H,CH),7.50-7.53(m,1H,CH),10.28(s,1H,NH),11.48(s,1H,NH)。
实施例17
化合物GDD-15:N-(正丙基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔正丙胺和20毫升1,2-二氯乙烷,搅拌至正丙胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,没有沉淀生成,在室温下搅拌4小时后再加热到80摄氏度条件下回流4小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:153-156摄氏度,收率56%;核磁数据(DMSO-d6,化学位移):0.89(t,3H,J=14.782Hz,CH3),1.49-1.54(m,2H,CH2),2.78(s,3H,CH3),3.15-3.20(m,2H,CH2),8.3(s,1H,NH),11.14(s,1H,NH)。
实施例18
化合物GDD-16:N-(苯并咪唑-2-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-胺基苯并咪唑和20毫升1,2-二氯乙烷,搅拌至邻硝基苯胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15 分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点大于250摄氏度,收率72%;核磁数据(DMSO-d6,化学位移):2.96(s,3H,CH3),7.13-7.25(m,1H,Ar-H),7.34-7.36(m,1H,Ar-H),7.53(d,J=8Hz,1H,Ar-H),7.62(d,J=8Hz,1H,Ar-H),8.33(s,1H,Ar-H),12.73(s,2H,NH)。
实施例19
化合物GDD-17:N-(3,5-二氯苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔3,5-二氯苯胺和20毫升1,2-二氯乙烷,搅拌至3,5-二氯苯胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:194-197摄氏度,收率72%;核磁数据(DMSO-d6,化学位移):2.82(s,3H,CH3),7.34(s,1H,CH),7.71(s,2H,CH),10.43(s,1H,NH),11.60(s,1H,NH)。
实施例20
化合物GDD-18:N-(4,6-二甲氧基嘧啶-2-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氨基-4,6-二甲氧基嘧啶和20毫升1,2-二氯乙烷,搅拌至2-氨基-4,6-二甲氧基嘧啶溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:184-187摄氏度,收率39%;核磁数据(DMSO-d6,化学位移):2.80(s,3H,CH3),3.82(s,6H,OMe),6.01(s,1H,CH),10.63(s,1H,NH),12.51(s,1H,NH)。
实施例21
化合物GDD-19:N-(4-溴苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔对溴苯胺和20毫升1,2-二氯乙烷,搅拌至对溴苯胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:195-197摄氏度,收率78%;核磁数据(DMSO-d6,化学位移):2.82(s,3H,CH3),7.55-7.56(m,4H,CH),10.29(s,1H,NH),11.49(s,1H,NH)。
实施例22
化合物GDD-20:N-(4-氯-6-甲氧基嘧啶-2-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成 及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氨基-4-氯-6-甲氧基嘧啶和20毫升1,2-二氯乙烷,搅拌至2-氨基-4-氯-6-甲氧基嘧啶溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:172-175摄氏度,收率63%;核磁数据(DMSO-d6,化学位移):2.82(s,3H,CH3),3.95(s,3H,OMe),6.88(s,1H,CH),10.88(s,1H,NH),11.79(s,1H,NH)。
实施例23
化合物GDD-21:N-(4,6-二甲基嘧啶-2-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氨基-4,6-二甲基嘧啶和20毫升1,2-二氯乙烷,搅拌至2-氨基-4,6-二甲基嘧啶溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点204-206摄氏度,收率21%;核磁数据(DMSO-d6,化学位移):2.40(s,6H,CH3),2.82(s,3H,CH3),7.01(s,1H,CH),10.64(s,1H,NH),12.71(s,1H,NH)。
实施例24
化合物GDD-22:N-(4-乙氧羰基-1,2,3-噻二唑-5-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔4-乙氧羰基-5-胺基-1,2,3-噻二唑和20毫升1,2-二氯乙烷,搅拌至4-乙氧羰基-5-胺基-1,2,3-噻二唑溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色晶体,熔点:163-165摄氏度,收率30%;核磁数据(DMSO,化学位移):1.42(t,J=6.4Hz,3H,CH3),2.85(s,1H,CH3),4.52(q,J=7.2Hz,J=14Hz,2H,CH2),12.58(s,1H,NH),12.74(s,1H,NH)。
实施例25
化合物GDD-23:N-(4,6-二氯嘧啶-2-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-胺基-4,6-二氯嘧啶和20毫升1,2-二氯乙烷,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固 体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色晶体,熔点:166-168摄氏度,收率63%;核磁数据(DMSO-d6,化学位移):2.83(s,3H,CH3),7.72(s,1H,Ar-H),11.14(s,1H,NH),11.67(s,1H,NH)。
实施例26
化合物GDD-24:N-(2,6-二氯嘧啶-4-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2,6-二氯-4-胺基嘧啶和20毫升1,2-二氯乙烷,搅拌至2-甲基-4-氯苯胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色晶体,熔点:173-175摄氏度,收率53%;核磁数据(DMSO-d6,化学位移):2.89(s,3H,CH3),6.51(s,1H,CH),11.17(s,1H,NH),11.90(s,1H,NH)。
实施例27
化合物GDD-25:N-(2-甲基-4-氯苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-甲基-4-氯苯胺和20毫升1,2-二氯乙烷,搅拌至2-甲基-4-氯苯胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤得固体,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色晶体,熔点:194-196摄氏度。核磁数据(DMSO-d6,化学位移):2.30(s,3H,CH3),2.83(s,1H,CH3),7.29(d,1H,J=8.738Hz,CH),7.38(s,1H,CH),7.96(d,1H,J=8.865Hz,CH),10.23(s,1H,NH),11.65(s,1H,NH)。
实施例28
化合物GDD-26:N-(2,6-二甲氧基嘧啶-4-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔4-氨基-2,6-二甲氧基嘧啶和20毫升1,2-二氯乙烷,搅拌至4-氨基2,6-二甲氧基嘧啶溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤得固体,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色晶体,熔点:191-193摄氏度,收率52%;核磁数据(DMSO-d6,化学位移):2.81(s,3H,CH3),3.89(d,6H,J=7.541Hz,OCH3),6.98(s,1H,CH),10.61(s,1H,NH),11.69(s,1H,NH)。
实施例29
化合物GDD-27:N-(3-氯苯基硫代甲酰胺基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔苯肼和20毫升1,2-二氯乙烷,搅拌至苯肼溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色晶体,熔点:167-169摄氏度。收率54%;核磁数据(DMSO-d6,化学位移):2.82(s,1H,CH3),3.91(s,1H,NH-CS),7.23(d,1H,J=7.327Hz,CH),7.36-7.39(m,1H,CH),7.47(d,1H,J=7.847Hz,CH),7.67(s,1H,CH),9.94(s,2H,NH),11.54(s,1H,NH)。
实施例30
化合物GDD-29:N-(7H-嘌呤-2-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-胺基-7H-嘌呤和20毫升1,2-二氯乙烷,搅拌至邻硝基苯胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点大于250摄氏度,收率45%;核磁数据(DMSO-d6,化学位移):2.84(s,1H,CH3),6.78(s,1H,NH),8.11(s,1H,Ar-H),8.58(s,1H,Ar-H),10.92(s,1H,NH),11.86(s,1H,NH)。
实施例31
化合物GDD-31:N-(5-氯吡啶-2-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氨基-5-氯吡啶和20毫升1,2-二氯乙烷,搅拌至2-氨基-5-氯吡啶完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色针状,熔点:198-199摄氏度,收率73%;核磁数据(DMSO-d6,化学位移):2.82(s,3H,CH3),7.98(d,1H,J=8.884Hz,CH),8.03(d,1H,J=8.809Hz,CH),8.41(s,1H,CH),10.72(s,1H,NH),11.72(s,1H,NH)。
实施例32
化合物GDD-32:N-(4-甲基-6-溴吡啶-2-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氨基-4-甲基-6-溴吡啶和20毫升1,2-二氯乙烷,搅拌至2-氨基-4-甲基-6-溴吡啶完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸 乙酯∶石油醚洗涤后干燥得产品;白色粉末,熔点:206-210摄氏度,收率54%;核磁数据(DMSO-d6,化学位移):2.39(s,3H,CH3C5H3NBr),2.82(s,3H,CH3),7.92(s,1H,CHCHN),8.43(s,1H,CHCBrN),10.70(s,1H,NH),11.71(s,1H,NH)。
实施例33
化合物GDD-33:N-(4-甲基吡啶-2-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氨基-4-甲基吡啶和20毫升1,2-二氯乙烷,搅拌至2-氨基-4-甲基吡啶完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色针状,熔点:207-209摄氏度,收率38%;核磁数据(DMSO-d6,化学位移):2.35(s,3H,CH3C5H3N),2.82(s,3H,CH3),7.01(d,1H,J=4.764Hz,CHCN),7.80(s,1H,CHCHN),8.20(d,1H,J=4.974Hz,CHN),10.62(s,1H,NH),11.72(s,1H,NH)。
实施例34
化合物GDD-34:N-环丙基甲基-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔环丙基甲胺和20毫升1,2-二氯乙烷,搅拌至环丙基甲胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色颗粒熔点:106-108摄氏度,收率23%;核磁数据(DMSO-d6,化学位移):0.012(m,2H,CH2),0.214(m,2H,CH2),0.794(s,1H,CH),2.542(s,3H,CH3),2.855(t,J=12.8Hz,2H,CH2),8.088(s,1H,NH),10.926(s,1H,NH)。
实施例35
化合物GDD-35:N-环丙基-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔环丙胺和20毫升1,2-二氯乙烷,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;熔点:138-140摄氏度,收率68%;核磁数据(DMSO-d6,化学位移):0.68-0.70(m,4H,C2H4),2.77(s,3H,CH3),7.56(s,1H,CH),7.63(s,1H,NH),11.03(s,1H,NH)。
实施例36化合物GDD-36:N-环丁基-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔环丁基胺和20毫升1,2-二氯乙烷,搅拌至溶解, 搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色颗粒熔点:129-131摄氏度,收率18%;核磁数据(DMSO-d6,化学位移):2.13(m,6H,CH2),2.77(s,3H,CH3),4.25(m,1H,CH),8.35(d,J=7.6Hz,1H,NH),8.92(s,1H,NH)。
实施例37
化合物GDD-37:N-(2-三氟甲基-5-氯苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-三氟甲基-5-氯苯胺和20毫升1,2-二氯乙烷,搅拌至2-三氟甲基-5-氯苯胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色颗粒熔点:190-192摄氏度,收率50%;核磁数据(DMSO-d6,化学位移):2.83(s,3H,CH3),7.54(d,1H,J=8.272Hz,CHCCl),7.84(d,1H,J=8.223Hz,CHCHCCl),8.66(s,1H,CHCCCF3),11.09(s,1H,NH),11.92(s,1H,NH)。
实施例38
化合物GDD-38:N-(2-氯-4-三氟甲基苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氯-4-三氟甲基苯胺和20毫升1,2-二氯乙烷,搅拌至2-氯-4-三氟甲基苯胺完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色针状,熔点:176-180摄氏度,收率23%;核磁数据(DMSO-d6,化学位移):2.83(s,3H,CH3),8.53(s,1H,CHCCF3),8.81(s,1H,CHN),11.13(s,1H,NH),11.86(s,1H,NH)。
实施例39
化合物GDD-39:N-(6-甲氧基苯并[d]噻唑-2-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氨基-6-甲氧基苯并[d]噻唑和20毫升1,2-二氯乙烷,搅拌至2-氨基-6-甲氧基苯并[d]噻唑溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色粉末,熔点:203-206摄氏度,收率55%;核磁数 据(DMSO-d6,化学位移):2.83(s,3H,CH3),3.82(s,3H,OCH3),7.05(d,1H,J=8.748Hz,CHCO),7.60(s,1H,CHCO),7.66(d,1H,J=8.870Hz,CHCCO),9.09(s,1H,NH),11.90(s,1H,NH)。
实施例40
化合物GDD-40:N-(6-甲基苯并[d]噻唑-2-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氨基-6-甲基苯并[d]噻唑和20毫升1,2-二氯乙烷,搅拌至2-氨基-6-甲基苯并[d]噻唑完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得白色晶体,熔点:189-192摄氏度,收率31%;核磁数据(DMSO-d6,化学位移):2.42(s,3H,CH3CCH),2.84(s,3H,CH3),7.39,(d,1H,,J=8.143Hz,CHCCH3),7.63-7.66(m,1H,CHCS),7.78(s,1H,CHCN),9.51(s,1H,NH),11.94(s,1H,NH)。
实施例41
化合物GDD-41:N-(4-甲基苯并[d]噻唑-2-基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氨基-4-甲基苯并[d]噻唑和20毫升1,2-二氯乙烷,搅拌至2-氨基-4-甲基苯并[d]噻唑完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色粉末,熔点:221-225摄氏度,收率48%;核磁数据(DMSO-d6,化学位移):2.58(s,3H,CH3CH),2.85(s,3H,CH3),7.20-7.32(m,2H,CHCH),7.81(d,1H,J=7.470Hz,CHCS),8.86(s,1H,NH),11.94(s,1H,NH)。
实施例42
化合物GDD-42:N-[(6-硝基)苯并[d]噻唑-2-基]-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氨基-6-硝基苯并[d]噻唑和20毫升1,2-二氯乙烷,搅拌至2-氨基-6-硝基苯并[d]噻唑完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后静置,抽滤得固体,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;黄色颗粒,熔点:208-210摄氏度,收率35%;核磁数据(DMSO-d6,化学位移):2.84(s,3H,CH3),7.94(s,1H,Ar-H),8.27(s,1H,Ar-H),8.30(s,1H,Ar-H),9.10(s,1H,NH),11.90(s,1H,NH)。
实施例43
化合物GDD-43:N-[(2-甲基)苯并[d]噻唑-6-基]-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成 及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔6-氨基-2-甲基苯并[d]噻唑和20毫升1,2-二氯乙烷,搅拌至6-氨基-2-甲基苯并[d]噻唑完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色晶体,熔点:210-212摄氏度,收率61%;核磁数据(DMSO-d6,化学位移):2.80(s,3H,CH3CS),2.84(s,3H,CH3),7.52(d,1H,J=8.581Hz,CH),7.98(d,1H,J=8.633Hz,CH),8.23(s,1H,CH),10.43(s,1H,NH),11.51(s,1H,NH)。
实施例44
化合物GDD-44:N-[(6-溴)苯并[d]噻唑-2-基]-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氨基-6-溴苯并[d]噻唑和20毫升1,2-二氯乙烷,搅拌至2-氨基-6-溴苯并[d]噻唑完全溶解,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离得固体产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色晶体,熔点:213-216摄氏度,收率35%;核磁数据(DMSO-d6,化学位移):2.84(s,3H,CH3),7.61(d,1H,J=8.629Hz,Ar-H),7.70(d,1H,J=8.568Hz,Ar-H),8.28(s,1H,CH),11.77(s,1H,NH),11.94(s,1H,NH)。
实施例45
化合物GDD-46:N-(2-氯苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔2-氯苯胺和20毫升1,2-二氯乙烷,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤得固体,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色晶体,熔点:171-173摄氏度,收率70%;核磁数据(DMSO-d6,化学位移):2.88(s,3H,CH3),7.23(t,J=7.6Hz,1H,Ar-H),7.45(t,J=8Hz,1H,Ar-H),7.62(d,J=8Hz,1H,Ar-H),8.33(d,J=8.4Hz,Ar-H),10.90(s,1H,NH),11.79(s,1H,NH)。
实施例46
化合物GDD-47:N-(4-氯苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔4-氯苯胺和20毫升1,2-二氯乙烷,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色晶体,熔点:173-175 摄氏度,收率72%;核磁数据(DMSO-d6,化学位移):2.83(s,3H,CH3),7.42(d,J=8Hz,1H,Ar-H),7.62(d,J=7.6Hz,1H,Ar-H),10.28(s,1H,NH),11.48(s,1H,NH)。
实施例47
化合物GDD-48:N-(3-硝基苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100毫升圆底烧瓶中加入2.7毫摩尔3-硝基苯胺和20毫升1,2-二氯乙烷,搅拌下滴加3毫摩尔4-甲基-1,2,3-噻二唑-5-甲酰基异氰酸酯,15分钟滴加完毕,立即产生沉淀,在室温下搅拌8小时,反应完毕后,静置,抽滤分离固体与溶剂,固体即为产物,滤液旋转蒸发除去溶剂,用体积比为1∶3的乙酸乙酯∶石油醚重结晶后放入冰箱中,析出固体,合并固体产物,用体积比为1∶3的乙酸乙酯∶石油醚洗涤后干燥得产品;白色晶体,熔点:171-173摄氏度,收率65%;核磁数据(DMSO-d6,化学位移):2.84(s,3H,CH3),7.66(t,J=8.2Hz,1H,Ar-H),7.96(m,2H,Ar-H),8.64(s,1H,Ar-H),10.54(s,1H,NH),11.60(s,1H,NH)。
实施例48
化合物GDD-49:N-(4-乙基苯基)-N’-(4-甲基-1,2,3-噻二唑-5-甲酰基)脲的合成及结构鉴定
在100
4-甲基-1,2,3-噻二唑-5-甲酰脲类化合物及其制备方法和用途专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。










动态评分
0.0