
配合物及其制备方法以及荧光纳米纤维的制备方法和应用")
IPC分类号 : C07J51/00,C09K11/06,G01N21/64









专利摘要
两亲性Tb(Ⅲ)配合物及其制备方法以及荧光纳米纤维的制备方法和应用,基于胆固醇分子的刚性骨架、生物相容性以及自组装等特点,设计制备了含胆固醇结构单元和双吡啶羧酸的两亲性Tb(Ⅲ)配合物,组装了基于此配合物的超分子荧光纳米纤维,研究了该荧光纳米纤维与金精三羧酸的作用机理,利用金精三羧酸与Tb(Ⅲ)离子间的协同络合以及能量转移过程实现了对金精三羧酸的高效传感。本发明所涉及的化学反应条件温和易实现,荧光纳米纤维的制备方法简便易操作,且所得纳米纤维尺寸均一,检测金精三羧酸的灵敏度高,选择性好,有望在研究金精三羧酸的药理过程中获得应用。
权利要求
1.一种两亲性Tb(Ⅲ)配合物,其特征在于,该配合物的结构式为:
。
2.根据权利要求1所述的两亲性Tb(Ⅲ)配合物的制备方法,其特征在于,包括以下步骤:
(1)合成式Ⅳ化合物:
在氮气保护条件下,向盛有乙腈的容器中加入式Ⅲ化合物、三乙胺、碘化钾、碳酸钾和6-氯甲基-2-甲酸乙酯吡啶,于81~90℃加热回流70~80h;过滤,旋蒸蒸除乙腈,通过柱色谱分离、重结晶,制得式Ⅳ化合物,其中,式Ⅲ化合物与三乙胺、碘化钾、碳酸钾、6-氯甲基-2-甲酸乙酯吡啶的摩尔比为1:1:(2~4):(4~8):(2~4),式Ⅲ化合物和乙腈的质量比为1:(50~100),式Ⅲ化合物、式Ⅳ化合物的结构式如下:
(2)合成式Ⅴ化合物:
将式Ⅳ化合物加入到盛有无水乙醇的圆底烧瓶中,搅拌下加热至50~80℃,待式Ⅳ化合物完全溶解后,冷却至室温,再加入氢氧化钠,于60~80℃下反应4~8h,旋蒸蒸除乙醇,逐滴滴加盐酸,产生白色 沉淀,抽滤,将所得固体真空干燥,制得式Ⅴ化合物,其中,式Ⅳ化合物与氢氧化钠的摩尔比为1:(4~6),式Ⅳ化合物与乙醇的质量比为1:(80~120),式Ⅴ化合物的结构式如下:
(3)合成式Ⅵ化合物:
将式Ⅴ化合物、氢氧化钠和pH值为7.4的HEPES缓冲溶液加入水中,加热至固体溶解,冷却至室温后,加入六水氯化铽并搅拌均匀,旋蒸抽滤,所得固体即为式Ⅵ化合物,其中,式Ⅴ化合物与HEPES、氢氧化钠摩尔比为1:10:(2~3),式Ⅴ化合物与水的质量比为1:(400~450),式Ⅴ化合物与六水氯化铽的摩尔比为1:1,式Ⅵ化合物的结构式如下:
。
3.根据权利要求2所述的一种两亲性Tb(Ⅲ)配合物的制备方法,其特征在于,式Ⅲ化合物通过以下步骤制得:
(1)合成6-羟甲基-2-甲酸乙酯吡啶:
在氮气保护条件下,将2,6-二甲酸乙酯吡啶和硼氢化钠加入到盛有无水乙醇的圆底烧瓶中,于90℃回流反应2~5h,调节pH至5,用三氯甲烷萃取,旋蒸蒸除三氯甲烷,以甲醇与三氯甲烷的体积比为1:15的混合液为流动相、硅胶为固定相进行柱色谱分离,制备成6-羟甲基-2-甲酸乙酯吡啶;其中,2,6-二甲酸乙酯吡啶和硼氢化钠的摩尔比是1:(0.5~0.7),2,6-二甲酸乙酯吡啶和无水乙醇的质量比1:(15~20);
(2)合成6-氯甲基-2-甲酸乙酯吡啶:
将6-羟甲基-2-甲酸乙酯吡啶和氯化亚砜加入到盛有无水二氯甲烷的圆底烧瓶中,冰浴下搅拌30min,然后室温下搅拌2~5h后,旋蒸蒸除二氯甲烷后用甲苯溶解,NaHCO3溶液洗涤,水洗有机层,旋蒸蒸除溶剂,制得6-氯甲基-2-甲酸乙酯吡啶,其中,6-羟甲基-2-甲酸乙酯吡啶和无水氯化亚砜的摩尔之比为1:(1~2);2,6-羟甲基-2-甲酸乙酯吡啶和二氯甲烷的质量之比为1:(20~50);
(3)合成式Ⅰ化合物:
室温条件下,将胆固醇、KOH溶液、十八冠六醚和丙烯腈依次加入到盛有二氯甲烷的圆底烧瓶中,室温反应8~12h后再加入三氯甲烷溶解、蒸馏水洗涤、旋蒸蒸除三氯甲烷,以乙酸乙酯与正己烷的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅰ化合物,其中,胆固醇与KOH、十八冠六醚和丙烯腈的摩尔比为1:(3~4):1:(2~4),胆固醇和二氯甲烷的质量比为1:(20~40),式Ⅰ化合物的结构式为:
(4)合成式Ⅱ化合物:
将式Ⅰ化合物、六水氯化镍和二碳酸二叔丁酯加入到盛有甲醇的圆底烧瓶中,室温搅拌15min后,加入硼氢化钠,室温反应12~24h后抽滤,旋蒸蒸除滤液,以乙酸乙酯与正己烷的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅱ化合物,其中,式Ⅰ化合物、六水氯化镍、二碳酸二叔丁酯和硼氢化钠的摩尔比为1:1:(1~3):(5~10),式Ⅰ化合物与甲醇的质量比为1:(120~150);式Ⅱ化合物的结构式如下:
(5)合成式Ⅲ化合物:
将式Ⅱ化合物与三氟乙酸加入到盛有二氯甲烷的圆底烧瓶中,室温搅拌2~8h,旋蒸蒸除二氯甲烷,以甲醇与三氯甲烷的体积比为1:10的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅲ化合物,其中,式Ⅱ化合物与三氟乙酸摩尔比为1:(20~30),三氟乙酸与二氯甲烷的体积比为1:4,式Ⅲ化合物的结构式如下:
。
4.根据权利要求2所述的两亲性Tb(Ⅲ)配合物的制备方法,其特征在于,所述步骤(1)合成式Ⅳ化合物中柱色谱分离采用四氢呋喃 与石油醚的体积为1:4的混合液为流动相、硅胶为固定相;
所述步骤(1)合成式Ⅳ化合物中重结晶采用正己烷作溶剂进行。
5.根据权利要求2所述的两亲性Tb(Ⅲ)配合物的制备方法,其特征在于,所述步骤(2)合成式Ⅴ化合物中盐酸的浓度为1mol/L。
6.根据权利要求3所述的两亲性Tb(Ⅲ)配合物的制备方法,其特征在于,所述步骤(3)合成式Ⅰ化合物中KOH溶液的质量分数为40%。
7.一种基于权利要求1所述的两亲性Tb(Ⅲ)配合物的荧光纳米纤维的制备方法,其特征在于,将HEPES溶于水中,调节pH值,配制成pH为7.4的HEPES缓冲溶液,然后将式化合物Ⅵ溶于10mmol/LpH值为7.4的HEPES缓冲溶液中,50~60℃加热1~3h后自然冷却至室温,得到25μmol/L的式Ⅵ化合物水溶液,即得到荧光纳米纤维。
8.根据权利要求7所述的荧光纳米纤维的制备方法,其特征在于,所述调节pH值是采用1mmol/L NaOH水溶液进行调节的。
9.一种如权利要求7所述的荧光纳米纤维在对金精三羧酸检测中的应用。
说明书
技术领域
本发明属于超分子传感材料技术领域,具体涉及两亲性Tb(Ⅲ)配合物及其制备方法以及荧光纳米纤维的制备方法和应用。
背景技术
金精三羧酸(Aurintricarboxylic acid,ATA)是一种芳香酸衍生物,可抑制细胞中蛋白与核酸之间的相互作用,进而抑制细胞的生长和分裂。例如:它被发现可以抑制甲型流感病毒的复制,也被证明能阻止艾滋病毒表面的gp120分子对T细胞CD4受体的入侵。因此,ATA有望被用于某些疾病的控制和治疗,而监测生命体中ATA的性能对于研究ATA的药理过程至关重要,因此研制性能优异的ATA传感器将会在很大程度上促进这一领域的发展。而要实现在生理条件对ATA分子的高效识别,要求传感器必须和阴离子间有强的相互作用力,并且能将这种识别过程转化放大成易检测的物理化学信号。
镧系金属配合物具有荧光寿命长、发射波长大且发射光谱带窄、等光学特征,近年来被广泛用于阴离子传感器的创制。镧系金属的电子f-f跃迁被禁阻,导致直接激发镧系金属非常困难,但当发色团(天线)与镧系金属离子间的距离足够小时,可通过激发发色团来间接激发镧系金属离子发出其特征荧光,此乃“turn on”型镧系金属配合物传感器的基本原理。据此,Valérie C.Pierre等人利用H2O2生成的羟基自由基与均苯三甲酸反应形成2-羟基-苯三甲酸,由于该化合物的羧基和相邻的羟基可与铽配合物发生协同络合而敏化铽的荧光,实现了对羟基自由基的检测。此外,该小组利用啡啶作为铽离子的能量给体,制备了一种基于铽配合物的荧光化学传感器,ATP、ADP与AMP与啡啶间的PET效应可阻断啡啶与铽的能量转移过程,进而猝灭铽的特征荧光,利用此三种核苷酸与铽配合物的络合能力不同实现了对ATP的选择性识别,并利用此原理实时监控了ATP水解酶对ATP的水解过程。尽管镧系金属配合物已被广泛用于阴离子的检测,但此类传感器的综合性能,如:选择性,灵敏度,仍需进一步提高,而且基于此类化合物的ATA传感器鲜有报道。因此制备性能优异的基于镧系金属配合物的ATA传感器不仅可以拓展阴离子传感器的研究领域,而且有利于深化对ATA参与的药理过程的理解。
不同于单分子荧光化学传感器,具有高级结构的超分子自组装体具备高度有序的纳米界面,且界面上的受体分子浓度远高于本体相,有利于协同络合多阴离子底物分子,从而实现对阴离子的高效传感。因此,本发明设计合成了两亲性Tb(Ⅲ)配合物,组装了基于此配合物的荧光纳米纤维,研究了该荧光纳米纤维与ATA的作用机理,利用ATA与Tb(Ⅲ)离子间的协同络合以及能量转移过程实现了对ATA的高效传感。
发明内容
本发明的目的在于克服现有技术中的问题,提供一种两亲性Tb(Ⅲ)配合物及其制备方法以及荧光纳米纤维的制备方法和应用,利用组装在纳米纤维表面的Tb(Ⅲ)配合物与阴离子间的协同络合作用实现对具有多个结合位点的阴离子的高选择性、高灵敏检测。
为实现上述目的,本发明采用如下的技术方案:
一种两亲性Tb(Ⅲ)配合物,该配合物的结构式为:
上述两亲性Tb(Ⅲ)配合物的制备方法,包括以下步骤:
(1)合成式Ⅳ化合物:
在氮气保护条件下,向盛有乙腈的容器中加入式Ⅲ化合物、三乙胺、碘化钾、碳酸钾和6-氯甲基-2-甲酸乙酯吡啶,于81~90℃加热回流70~80h;过滤,旋蒸蒸除乙腈,通过柱色谱分离、重结晶,制得式Ⅳ化合物,其中,式Ⅲ化合物与三乙胺、碘化钾、碳酸钾、6-氯甲基-2-甲酸乙酯吡啶的摩尔比为1:1:(2~4):(4~8):(2~4),式Ⅲ化合物和乙腈的质量比为1:(50~100),式Ⅲ化合物、式Ⅳ化合物的结构式如下:
(2)合成式Ⅴ化合物:
将式Ⅳ化合物加入到盛有无水乙醇的圆底烧瓶中,搅拌下加热至50~80℃,恒温反应1~2h,自然冷却至室温,再加入氢氧化钠,于60~80℃下加热4~8h,然后旋蒸蒸除无水乙醇,逐滴滴加盐酸,产生白色沉淀,抽滤,将所得固体真空干燥,制得式Ⅴ化合物,其中,式Ⅳ化合物与氢氧化钠的摩尔比为1:(4~6),式Ⅳ化合物与乙醇的质量比为1:(80~120),式Ⅴ化合物的结构式如下:
(3)合成式Ⅵ化合物:
将式Ⅴ化合物、氢氧化钠和pH值为7.4的HEPES缓冲溶液加入水中,加热至固体溶解,冷却至室温后,加入六水氯化铽并搅拌均匀,旋蒸抽滤,所得固体即为式Ⅵ化合物,其中,式Ⅴ化合物与HEPES、氢氧化钠摩尔比为1:10:(2~3),式Ⅴ化合物与水的质量比为1:(400~450),式Ⅴ化合物与六水氯化铽的摩尔比为1:1,式Ⅵ化合物的结构式如下:
式Ⅲ化合物通过以下步骤制得:
(1)合成6-羟甲基-2-甲酸乙酯吡啶:
在氮气保护条件下,将2,6-二甲酸乙酯吡啶和硼氢化钠加入到盛有无水乙醇的圆底烧瓶中,90℃回流反应2~5h,然后调节pH至5,用三氯甲烷萃取,旋蒸蒸除三氯甲烷后以甲醇与三氯甲烷的体积比为1:15的混合液为流动相、硅胶为固定相进行柱色谱分离,制备成6-羟甲基-2-甲酸乙酯吡啶;其中,2,6-二甲酸乙酯吡啶和硼氢化钠的摩尔比是1:(0.5~0.7),2,6-二甲酸乙酯吡啶和无水乙醇的质量比1:(15~20);
(2)合成6-氯甲基-2-甲酸乙酯吡啶:
将6-羟甲基-2-甲酸乙酯吡啶和氯化亚砜加入到盛有无水二氯甲烷的圆底烧瓶中,冰浴下搅拌30min,然后室温下搅拌2~5h后,旋蒸蒸除二氯甲烷后用甲苯溶解,NaHCO3溶液洗涤,水洗有机层,旋蒸蒸除溶剂,制得6-氯甲基-2-甲酸乙酯吡啶,其中,6-羟甲基-2-甲酸乙酯吡啶和无水氯化亚砜的摩尔之比为1:(1~2);2,6-羟甲基-2-甲酸乙酯吡啶和二氯甲烷的质量之比为1:(20~50);
(3)合成式Ⅰ化合物:
室温条件下,将胆固醇、KOH溶液、十八冠六醚和丙烯腈依次加入到盛有无水二氯甲烷的中,室温反应8~12h后再加入三氯甲烷溶解、蒸馏水洗涤、旋蒸蒸除三氯甲烷后以乙酸乙酯与正己烷的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅰ化合物,其中,胆固醇与KOH溶液、十八冠六醚和丙烯腈的摩尔比为1:(3~4):1:(2~4),胆固醇和二氯甲烷的质量比为1:(20~40),式Ⅰ化合物的结构式为:
(4)合成式Ⅱ化合物:
将式Ⅰ化合物、六水氯化镍和二碳酸二叔丁酯加入到盛有无水甲醇的圆底烧瓶中,室温搅拌15min后,再加入硼氢化钠,室温反应12~24h后抽滤,旋蒸蒸除溶剂,以乙酸乙酯与正己烷的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅱ化合物,其中,式Ⅰ化合物、六水氯化镍、二碳酸二叔丁酯和硼氢化钠的摩尔比为1:1:(1~3):(5~10),式Ⅰ化合物与无水甲醇的质量比为1:(120~150);式Ⅱ化合物的结构式如下:
(5)合成式Ⅲ化合物:
将式Ⅱ化合物与三氟乙酸加入到盛有无水二氯甲烷的圆底烧瓶中,室温搅拌2~8h后旋蒸蒸除二氯甲烷,以甲醇与三氯甲烷的体积比为1:10的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅲ化合物,其中,式Ⅱ化合物与三氟乙酸摩尔比为1:(20~30),三氟乙酸与二氯甲烷的体积比为1:4,式Ⅲ化合物的结构式如下:
所述步骤(1)合成式Ⅳ化合物中柱色谱分离采用四氢呋喃与石油醚的体积为1:4的混合液为流动相、硅胶为固定相;
所述步骤(1)合成式Ⅳ化合物中重结晶采用正己烷作溶剂进行。
所述步骤(2)合成式Ⅴ化合物中盐酸的浓度为1mol/L。
所述步骤(3)合成式Ⅰ化合物中KOH溶液的质量分数为40%。
一种基于两亲性Tb(Ⅲ)配合物的荧光纳米纤维的制备方法,将HEPES溶于水中,调节pH值,配制成pH为7.4的HEPES缓冲溶液,然后将式化合物Ⅵ溶于10mmol/L pH值为7.4的HEPES缓冲溶液中,50~60℃加热1~3h后自然冷却至室温,得到25μmol/L的式Ⅵ化合物水溶液,即得到荧光纳米纤维。
所述调节pH值是采用1mmol/L NaOH水溶液进行调节的。
一种荧光纳米纤维在对金精三羧酸检测中的应用。
与现有技术相比,本发明具有的有益效果:本发明利用胆固醇具有超分子自组装、刚性结构和生物相容性的特点,将其作为两亲性Tb(Ⅲ)配合物的疏水基团,制得含有胆固醇结构单元和双吡啶羧酸的两亲性Tb(Ⅲ)配合物,制备方法简单,操作简便,反应条件温和。
利用Tb(Ⅲ)配合物具有荧光寿命长、斯托克斯位移大和配位不饱和的特点,将其作为亲水基团,该两亲性Tb(Ⅲ)配合物在水相中自组装形成纳米纤维型超分子传感界面,将这一新型纳米纤维作为传感平台,利用高度有序的超分子传感界面具有预组织结构、界面上的分子浓度远高于本体相,有利于协同络合具有多个结合位点的阴离子的特点,同时金精三羧酸(ATA)作为Tb(Ⅲ)离子的能量给体,可协同络合纳米纤维表面的Tb(Ⅲ)离子而敏化其荧光,从而实现对ATA的高效传感。本发明所制备的荧光纳米纤维,结构均一,荧光寿命长、检测ATA的灵敏度高,为深化认识ATA参与的药理过程奠定了一定的基础。
附图说明
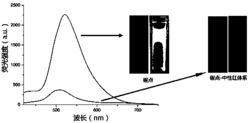
图1是实施例1制备的含胆固醇结构单元和双吡啶羧酸的两亲性Tb(Ⅲ)配合物的透射电镜图。
图2是实施例1制备的含胆固醇结构单元和双吡啶羧酸的两亲性Tb(Ⅲ)配合物的荧光发射光谱图。
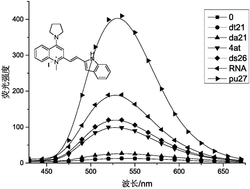
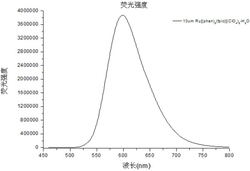
图3是实施例1制备的含胆固醇结构单元和双吡啶羧酸的两亲性Tb(Ⅲ)配合物在不同ATA浓度时的荧光光谱图。
具体实施方式
下面结合附图和实施例对本发明进一步详细说明,但本发明不限于这些实施例。本发明中无水二氯甲烷中的水分小于0.1%,无水乙醇中的水分小于0.5%。
本发明中两亲性Tb(Ⅲ)配合物的结构式为:
下面通过具体实施例说明上述两亲性Tb(Ⅲ)配合物的制备方法。
实施例1
(1)合成6-羟甲基-2-甲酸乙酯吡啶
在流速为1mL/s的氮气条件下,将2,6-二甲酸乙酯吡啶5g和硼氢化钠0.424g加入到盛有95mL无水乙醇的圆底烧瓶中,2,6-二甲酸乙酯吡啶和硼氢化钠的摩尔比是1:0.5。于90℃回流反应5h,然后调节pH值至5,用三氯甲烷萃取,无水硫酸钠干燥,旋蒸蒸除三氯甲烷后以甲醇与三氯甲烷的体积比为1:15的混合液为流动相、硅胶为固定相进行柱色谱分离,得到6-羟甲基-2-甲酸乙酯吡啶。
1H-NMR,δH(400MHz,CDCl3,Me4Si):1.43(3H,O-CH2-CH3),4.47(2H,O-CH2-CH3),4.87(2H,CH2-OH),7.52(1H,pyridine protons),7.85(1H,pyridine protons),8.03(1H,pyridine protons)。其反应方程如下:
(2)合成6-氯甲基-2-甲酸乙酯吡啶
将6-羟甲基-2-甲酸乙酯吡啶1.81g和氯化亚砜730μL加入到盛有28mL无水二氯甲烷的圆底烧瓶中,6-羟甲基-2-甲酸乙酯吡啶和氯化亚砜的摩尔之比为1:1,冰浴下搅拌30min,然后室温搅拌5h。反应结束后,旋蒸蒸除二氯甲烷,甲苯溶解、0.1mol/L碳酸氢钠溶液洗涤,然后水洗有机层至中性,无水硫酸钠干燥后旋蒸蒸除溶剂,得到6-氯甲基-2-甲酸乙酯吡啶。
1H NMR,δH(400MHz,CDCl3,Me4Si):8.08(1H,pyridine protons),7.91(1H,pyridine protons),7.74(1H,pyridine protons),4.79(2H,CH2-Cl),4.52(2H,O-CH2-CH3),1.46(3H,O-CH2-CH3)。其反应方程如下:
(3)合成式Ⅰ化合物
室温条件下,将胆固醇3.088g、质量分数为40%的KOH溶液1.3mL、十八冠六醚0.208g和丙烯腈1.05mL依次加入到盛有46mL无水二氯甲烷的圆底烧瓶中,胆固醇与KOH溶液、十八冠六醚和丙烯腈的摩尔比为1:3:1:2,室温搅拌12h,蒸馏水洗涤、干燥除水,旋蒸蒸除溶剂后以乙酸乙酯与正己烷的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,得到化合式Ⅰ化合物,1H-NMR,δH(400MHz,CDCl3,Me4Si):5.35(1H,alkenyl),3.67(2H,NC-CH2-CH2-O),3.22(1H,oxycyclohexyl),2.58(2H,NC-CH2-CH2-O),0.67-2.21(42H,cholesteryl protons)。其反应方程如下:
(4)合成式Ⅱ化合物
将式Ⅰ化合物2.916g、六水氯化镍1.56g和二碳酸二叔丁酯1.435g加入到盛有110mL无水甲醇的圆底烧瓶中,室温搅拌5min后,再缓慢加入硼氢化钠1.221g,式Ⅰ化合物、六水氯化镍、二碳酸二叔丁酯和硼氢化钠的摩尔比为1:1:1:5,室温反应24h,抽滤,旋蒸蒸除滤液后,以乙酸乙酯与正己烷的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅱ化合物,1H-NMR,δH(400MHz,CDCl3,Me4Si):1.44(9H,(CH3)3COCONH),1.7(2H,Boc-NH-CH2-CH2-CH2-O),3.13(1H,oxycyclohexyl),3.22(2H,NHCH2),3.53(2H,CH2O),4.92(1H,NH),5.34(1H,alkenyl),0.67-2.21(42H,cholesteryl protons)。其反应方程式如下:
(5)合成式Ⅲ化合物
将式Ⅱ化合物1.939g加入到盛有6mL无水二氯甲烷的圆底烧瓶中,然后加入三氟乙酸24mL,式Ⅱ化合物与三氟乙酸摩尔比为1:20,三氟乙酸与无水二氯甲烷的体积比为1:4,室温搅拌2h,旋蒸蒸除二氯甲烷后以甲醇与三氯甲烷的体积比为1:10的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅲ化合物,1H-NMR,δH(400MHz,CDCl3,Me4Si):3.05(3H,oxycyclohexyl+2H(O-CH2)),3.58(2H,CH2NH3+),5.24(1H,alkenyl),7.99(3H,NH3+),0.67-2.21(42H,cholesteryl protons)。其反应方程式如下:
(6)合成式Ⅳ化合物
在流速为1mL/s的氮气条件下,将盛有75mL乙腈的150mL三颈烧瓶中加入式Ⅲ化合物1.170g、三乙胺293μL、碘化钾0.697g、碳酸钾1.159g和6-氯甲基-2-甲酸乙酯吡啶0.835g,于90℃加热回流72h。式Ⅲ化合物与三乙胺、碘化钾、碳酸钾、6-氯甲基-2-甲酸乙酯吡啶的摩尔比为1:1:2:4:2,式Ⅲ化合物和乙腈的质量比为1:50。过滤,将滤液旋蒸蒸除后以四氢呋喃与石油醚的体积为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,用甲醇共旋,35℃真空干燥后,用正己烷重结晶、离心,得到式Ⅳ化合物,1H-NMR,δH(400MHz,CDCl3,Me4Si):7.8(6H,pyridine protons),5.3(1H,alkenyl),4.45(4H,-O-CH2-CH3),3.94(4H,-N-CH2-C),3.46(2H,O-CH2-CH2-CH2-N),3.05(1H,oxycyclohexyl),2.66(2H,O-CH2-CH2-CH2-N),1.43(6H,O-CH2-CH3),0.67-2.21(42H,cholesteryl protons)。其反应方程式如下:
(7)合成式Ⅴ化合物
将式Ⅳ化合物0.243g加入到盛有24mL无水乙醇的圆底烧瓶中,加热至50℃,恒温搅拌2h,自然冷却至室温,加入0.5mol/L的氢氧化钠水溶液2.52mL,式Ⅳ化合物与氢氧化钠的摩尔比为1:4,式Ⅳ化合物与乙醇的质量比为1:80,60℃条件下加热5h,然后旋蒸蒸除溶液至2.52mL,再逐滴滴加1mol/L的盐酸,产生白色沉淀,抽滤、真空干燥,得到式Ⅴ化合物,1H-NMR,δH(400MHz,CDCl3,Me4Si):8.14(1H,pyridine protons),8.04(1H,pyridine protons),7.74(1H,pyridine protons),5.20(1H,alkenyl),4.81(4H,N-CH2-C),3.66(2H,O-CH2-CH2-CH2-N),3.10(1H,oxycyclohexyl),2.18(2H,O-CH2-CH2-CH2-N),0.67-2.21(42H,cholesteryl protons)。其反应方程式如下:
(8)合成式Ⅵ化合物
将式Ⅴ化合物0.022g加入8.8mL水中,再依次加入200mmol/L缓冲溶液(pH=7.4)500μL和200mmol/L氢氧化钠溶液300μL,式Ⅴ化合物与HEPES(4-羟乙基哌嗪乙磺酸)、氢氧化钠摩尔比为1:10:2,式Ⅴ化合物与水的质量比为1:400,50℃条件下加热至固体溶解,冷却至室温后,缓慢加入10mmol/L六水氯化铽水溶液3mL,式Ⅴ化合物与六水氯化铽的摩尔比为1:1,旋蒸抽滤,所得固体即为式Ⅵ化合物,MS(m/z,ESI+)C48H71N3O5,m/z=870.3910,IR:vmax(KBr,cm-1)3071(C-H),1638(C=O),1582(C=N),1150(C-N),1092(C-O)。其反应方程式如下:
基于两亲性Tb(Ⅲ)配合物的荧光纳米纤维的制备方法:将HEPES溶于水中,通过1mmol/L的氢氧化钠水溶液调节pH值,配制成pH为7.4、10mmol/L的HEPES缓冲溶液,然后将式化合物Ⅵ溶于10mmol/L pH值为7.4的HEPES缓冲溶液中,50℃加热1h后自然冷却至室温,得到25μmol/L的式Ⅵ化合物水溶液,即得到荧光纳米纤维。
实施例2
(1)合成6-羟甲基-2-甲酸乙酯吡啶
在流速为1mL/s的氮气条件下,将2,6-二甲酸乙酯吡啶5g和硼氢化钠0.505g加入到盛有125mL无水乙醇的圆底烧瓶中,2,6-二甲酸乙酯吡啶和硼氢化钠的摩尔比是1:0.6,90℃回流反应5h,然后调节pH至5左右,用三氯甲烷萃取,无水硫酸钠干燥,旋蒸蒸除三氯甲烷后以甲醇与三氯甲烷的体积比为1:15的混合液为流动相、硅胶为固定相进行柱色谱分离,得到6-羟甲基-2-甲酸乙酯吡啶。1H-NMR,δH(400MHz,CDCl3,Me4Si):1.43(3H,O-CH2-CH3),4.47(2H,O-CH2-CH3),4.87(2H,CH2-OH),7.52(1H,pyridine protons),7.85(1H,pyridine protons),8.03(1H,pyridine protons)。
(2)合成6-氯甲基-2-甲酸乙酯吡啶
将6-羟甲基-2-甲酸乙酯吡啶1.81g和氯化亚砜876μL加入到盛有40mL无水二氯甲烷的圆底烧瓶中,6-羟甲基-2-甲酸乙酯吡啶和氯化亚砜的摩尔之比为1:1.2,冰浴下搅拌30min,然后室温搅拌5h。反应结束后,旋蒸蒸除二氯甲烷,甲苯溶解、0.1mol/L碳酸氢钠溶液洗涤,然后水洗有机层至中性,无水硫酸钠干燥后旋蒸蒸除溶剂,得到6-氯甲基-2-甲酸乙酯吡啶,1H NMR,δH(400MHz,CDCl3,Me4Si):8.08(1H,pyridine protons),7.91(1H,pyridine protons),7.74(1H,pyridine protons),4.79(2H,CH2-Cl),4.52(2H,O-CH2-CH3),1.46(3H,O-CH2-CH3)。
(3)合成式Ⅰ化合物
室温条件下,将胆固醇3.088g、质量分数为40%的KOH溶液1.6mL、十八冠六醚0.208g和丙烯腈1.2mL依次加入到盛有60mL无水二氯甲烷的圆底烧瓶中,胆固醇与KOH、十八冠六醚和丙烯腈的摩尔比为1:3.3:1:4,室温搅拌12h,蒸馏水洗涤、干燥除水,旋蒸蒸除溶剂后以乙酸乙酯与正己烷的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,得到化合式Ⅰ化合物,1H-NMR,δH(400MHz,CDCl3,Me4Si):5.35(1H,alkenyl),3.67(2H,NC-CH2-CH2-O),3.22(1H,oxycyclohexyl),2.58(2H,NC-CH2-CH2-O),0.67-2.21(42H,cholesteryl protons)。其反应方程如下:
(4)合成式Ⅱ化合物
将式Ⅰ化合物2.916g、六水氯化镍1.56g和二碳酸二叔丁酯1.722g加入到盛有120mL无水甲醇的圆底烧瓶中,室温搅拌5min后,再缓慢加入硼氢化钠1.710g,式Ⅰ化合物、六水氯化镍、二碳酸二叔丁酯和硼氢化钠的摩尔比为1:1:1.2:7,室温反应24h,抽滤,旋蒸蒸除滤液后以乙酸乙酯与正己烷的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,得到成式Ⅱ化合物,1H-NMR,δH(400MHz,CDCl3,Me4Si):1.44(9H,(CH3)3COCONH),1.7(2H,Boc-NH-CH2-CH2-CH2-O),3.13(1H,oxycyclohexyl),3.22(2H,NHCH2),3.53(2H,CH2O),4.92(1H,NH),5.34(1H,alkenyl),0.67-2.21(42H,cholesteryl protons)。
(5)合成式Ⅲ化合物
将式Ⅱ化合物1.939g加入到盛有28mL无水二氯甲烷的圆底烧瓶中,然后加入三氟乙酸7mL,三氟乙酸与无水二氯甲烷的体积比为1:4,室温搅拌2h,旋蒸蒸除二氯甲烷后以甲醇与三氯甲烷的体积比为1:10的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅲ化合物,1H-NMR,δH(400MHz,CDCl3,Me4Si):3.05(3H,oxycyclohexyl+2H(O-CH2)),3.58(2H,CH2NH3+),5.24(1H,alkenyl),7.99(3H,NH3+),0.67-2.21(42H,cholesteryl protons)。
(6)合成式Ⅳ化合物
在流速为1.2mL/s的氮气条件下,将盛有80mL乙腈的150mL三颈烧瓶中加入式Ⅲ化合物1.170g、三乙胺293μL、碘化钾1.046g、碳酸钾1.739g和6-氯甲基-2-甲酸乙酯吡啶1.253g,90℃加热回流72h。式Ⅲ化合物与三乙胺、碘化钾、碳酸钾、6-氯甲基-2-甲酸乙酯吡啶的摩尔比为1:1:3:6:3,式Ⅲ化合物和乙腈的质量比为1:54。过滤,将滤液旋蒸蒸除后以四氢呋喃与石油醚的体积为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,用甲醇共旋,35℃真空干燥后,用正己烷重结晶、离心,得到式Ⅳ化合物,1H-NMR,δH(400MHz,CDCl3,Me4Si):7.8(6H,pyridine protons),5.3(1H,alkenyl),4.45(4H,-O-CH2-CH3),3.94(4H,-N-CH2-C),3.46(2H,O-CH2-CH2-CH2-N),3.05(1H,oxycyclohexyl),2.66(2H,O-CH2-CH2-CH2-N),1.43(6H,O-CH2-CH3),0.67-2.21(42H,cholesteryl protons)。
(7)合成式Ⅴ化合物
将式Ⅳ化合物0.243g加入到盛有30mL无水乙醇的圆底烧瓶中,加热至50℃,恒温搅拌2h,自然冷却至室温,然后加入0.5mol/L氢氧化钠溶液3.78mL,式Ⅳ化合物与氢氧化钠的摩尔比为1:6,式Ⅳ化合物与乙醇的质量比为1:98,60℃条件下加热5h,然后旋蒸蒸除溶液至3.78mL,再逐滴滴加1mol/L的盐酸,产生白色沉淀,抽滤、真空干燥,得到式Ⅴ化合物,1H-NMR,δH(400MHz,CDCl3,Me4Si):8.14(1H,pyridine protons),8.04(1H,pyridine protons),7.74(1H,pyridine protons),5.20(1H,alkenyl),4.81(4H,N-CH2-C),3.66(2H,O-CH2-CH2-CH2-N),3.10(1H,oxycyclohexyl),2.18(2H,O-CH2-CH2-CH2-N),0.67-2.21(42H,cholesteryl protons)。
(8)合成式Ⅵ化合物
将式Ⅴ化合物0.022g加入到盛有8.975mL水的圆底烧瓶中,再依次加入200mmol/L氢氧化钠溶液525μL和200mmol/L缓冲溶液(pH=7.4)450μL,式Ⅴ化合物与HEPES、氢氧化钠摩尔比为1:10:3,式Ⅴ化合物与水的质量比为1:450,50℃条件下加热至固体溶解,冷却至室温后,缓慢加入10mmol/L六水氯化铽水溶液3mL,式Ⅴ化合物与六水氯化铽的摩尔比为1:1,旋蒸抽滤,所得固体即为式Ⅵ化合物,MS(m/z,ESI+)C48H71N3O5,m/z=870.3910。IR:vmax(KBr,cm-1)3071(C-H),1638(C=O),1582(C=N),1150(C-N),1092(C-O)。
基于两亲性Tb(Ⅲ)配合物的荧光纳米纤维的制备方法:将HEPES溶于水中,通过1mmol/L的氢氧化钠水溶液调节pH值,配制成pH为7.4的HEPES缓冲溶液,然后将式化合物Ⅵ溶于10mmol/L pH值为7.4的HEPES缓冲溶液中,60℃加热1h后自然冷却至室温,得到25μmol/L的式Ⅵ化合物水溶液,即得到荧光纳米纤维。
实施例3
(1)合成6-羟甲基-2-甲酸乙酯吡啶
在流速为1.2mL/s的氮气条件下,将2,6-二甲酸乙酯吡啶5g和硼氢化钠0.509g加入到盛有130mL无水乙醇的圆底烧瓶中,2,6-二甲酸乙酯吡啶和硼氢化钠的摩尔比是1:0.7。90℃回流反应5h,然后调节pH至5,用三氯甲烷萃取,无水硫酸钠干燥,旋蒸蒸除三氯甲烷后以甲醇与三氯甲烷的体积比为1:15的混合液为流动相、硅胶为固定相进行柱色谱分离,得到6-羟甲基-2-甲酸乙酯吡啶。1H-NMR,δH(400MHz,CDCl3,Me4Si):1.43(3H,O-CH2-CH3),4.47(2H,O-CH2-CH3),4.87(2H,CH2-OH),7.52(1H,pyridine protons),7.85(1H,pyridine protons),8.03(1H,pyridine protons)。
(2)合成6-氯甲基-2-甲酸乙酯吡啶
将6-羟甲基-2-甲酸乙酯吡啶1.81g和氯化亚砜1460μL加入到盛有68mL无水二氯甲烷的圆底烧瓶中,6-羟甲基-2-甲酸乙酯吡啶和氯化亚砜的摩尔之比为1:2,冰浴下搅拌30min,然后室温搅拌5h。反应结束后,旋蒸蒸除二氯甲烷,甲苯溶解、0.1mol/L碳酸氢钠溶液洗涤,然后水洗有机层至中性,无水硫酸钠干燥后旋蒸蒸除溶剂,得到6-氯甲基-2-甲酸乙酯吡啶,1H NMR,δH(400MHz,CDCl3,Me4Si):8.08(1H,pyridine protons),7.91(1H,pyridine protons),7.74(1H,pyridine protons),4.79(2H,CH2-Cl),4.52(2H,O-CH2-CH3),1.46(3H,O-CH2-CH3)。
(3)合成式Ⅰ化合物
室温条件下,将胆固醇3.088g、质量分数为40%的KOH溶液2mL、十八冠六醚0.208g和丙烯腈2.1mL依次加入到盛有92mL无水二氯甲烷的圆底烧瓶中,胆固醇与KOH、十八冠六醚和丙烯腈的摩尔比为1:4:1:4,室温搅拌12h,蒸馏水洗涤、干燥除水,旋蒸蒸除溶剂后以乙酸乙酯与正己烷的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,得到化合式Ⅰ化合物,1H-NMR,δH(400MHz,CDCl3,Me4Si):5.35(1H,alkenyl),3.67(2H,NC-CH2-CH2-O),3.22(1H,oxycyclohexyl),2.58(2H,NC-CH2-CH2-O),0.67-2.21(42H,cholesteryl protons)。
(4)合成式Ⅱ化合物
将式Ⅰ化合物2.916g、六水氯化镍1.56g和二碳酸二叔丁酯4.305g加入到盛有130mL无水甲醇的圆底烧瓶中,室温搅拌5min后,再缓慢加入硼氢化钠2.442g,式Ⅰ化合物、六水氯化镍、二碳酸二叔丁酯和硼氢化钠的摩尔比为1:1:2:10,室温反应24h,抽滤,旋蒸蒸除滤液后以乙酸乙酯与正己烷的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅱ化合物,1H-NMR,δH(400MHz,CDCl3,Me4Si):1.44(9H,(CH3)3COCONH),1.7(2H,Boc-NH-CH2-CH2-CH2-O),3.13(1H,oxycyclohexyl),3.22(2H,NHCH2),3.53(2H,CH2O),4.92(1H,NH),5.34(1H,alkenyl),0.67-2.21(42H,cholesteryl protons)。
(5)合成式Ⅲ化合物
将式Ⅱ化合物1.939g加入到盛有36mL无水二氯甲烷的圆底烧瓶中,然后加入三氟乙酸9mL,三氟乙酸与无水二氯甲烷的体积比为1:4,室温搅拌2h,旋蒸蒸除二氯甲烷后以甲醇与三氯甲烷的体积比为1:10的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅲ化合物,1H-NMR,δH(400MHz,CDCl3,Me4Si):3.05(3H,oxycyclohexyl+2H(O-CH2)),3.58(2H,CH2NH3+),5.24(1H,alkenyl),7.99(3H,NH3+),0.67-2.21(42H,cholesteryl protons)。
(6)合成式Ⅳ化合物
在流速为1.2mL/s的氮气条件下,在盛有148mL乙腈的150mL三颈烧瓶中加入式Ⅲ化合物1.170g、三乙胺293μL、碘化钾1.394g、碳酸钾2.318g和6-氯甲基-2-甲酸乙酯吡啶1.67g,90℃加热回流72h,然后过滤,将滤液旋蒸蒸除后以四氢呋喃与石油醚的体积为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,用甲醇共旋,35℃真空干燥后,用正己烷重结晶、离心,得到式Ⅳ化合物,其中,式Ⅲ化合物与三乙胺、碘化钾、碳酸钾、6-氯甲基-2-甲酸乙酯吡啶的摩尔比为1:1:4:8:4,式Ⅲ化合物和乙腈的质量比为1:100。1H-NMR,δH(400MHz,CDCl3,Me4Si):7.8(6H,pyridine protons),5.3(1H,alkenyl),4.45(4H,-O-CH2-CH3),3.94(4H,-N-CH2-C),3.46(2H,O-CH2-CH2-CH2-N),3.05(1H,oxycyclohexyl),2.66(2H,O-CH2-CH2-CH2-N),1.43(6H,O-CH2-CH3),0.67-2.21(42H,cholesteryl protons)。
(7)合成式Ⅴ化合物
将式Ⅳ化合物0.243g加入到盛有37mL无水乙醇的圆底烧瓶中,加热至50℃,恒温搅拌2h,自然冷却至室温,加入0.5mol/L的氢氧化钠溶液3.15mL,式Ⅳ化合物与氢氧化钠的摩尔比为1:5,式Ⅳ化合物与乙醇的质量比为1:120,60℃条件下加热5h,然后旋蒸蒸除溶液至3.15mL,再逐滴滴加1mol/L的盐酸,产生白色沉淀,抽滤、真空干燥,得到式Ⅴ化合物,1H-NMR,δH(400MHz,CDCl3,Me4Si):8.14(1H,pyridine protons),8.04(1H,pyridine protons),7.74(1H,pyridine protons),5.20(1H,alkenyl),4.81(4H,N-CH2-C),3.66(2H,O-CH2-CH2-CH2-N),3.10(1H,oxycyclohexyl),2.18(2H,O-CH2-CH2-CH2-N),0.67-2.21(42H,cholesteryl protons)。
(8)合成式Ⅵ化合物
将式Ⅴ化合物0.022g加入8.975mL水中,再依次加入200mmol/L氢氧化钠溶液525μL和200mmol/L缓冲溶液(pH=7.4)500μL,式Ⅴ化合物与HEPES、氢氧化钠摩尔比为1:10:2.5,式Ⅴ化合物与水的质量比为1:450,80℃条件下加热至固体溶解,冷却至室温后,缓慢加入10mmol/L六水氯化铽水溶液3mL,式Ⅴ化合物与六水氯化铽的摩尔比为1:1,旋蒸抽滤,所得固体即为式Ⅵ化合物,MS(m/z,ESI+)C48H71N3O5,m/z=870.3910。IR:vmax(KBr,cm-1)3071(C-H),1638(C=O),1582(C=N),1150(C-N),1092(C-O)。
基于两亲性Tb(Ⅲ)配合物的荧光纳米纤维的制备方法:将HEPES溶于水中,通过1mmol/L的氢氧化钠水溶液调节pH值,配制成pH为7.4的HEPES缓冲溶液,然后将式化合物Ⅵ溶于10mmol/L pH值为7.4的HEPES缓冲溶液中,55℃加热2h后自然冷却至室温,得到25μmol/L的式Ⅵ化合物水溶液,即得到荧光纳米纤维。
所制备的荧光纳米纤维的形貌及光物理性质分别采用透射电镜、FLS920型单光子计数时间分辨荧光光谱仪进行表征,结果见图1和图2。
由图1可见,所制备含胆固醇结构单元和双吡啶羧酸的两亲性Tb(Ⅲ)配合物的超分子自组装体是尺寸均一的纳米纤维状,其直径为10nm,长度可达微米级。由图2可见,含胆固醇结构单元和双吡啶羧酸的两亲性Tb(Ⅲ)配合物在268nm激发波长下发射出铽的特征光,表明配体和Tb3+形成了稳定配合物。
基于实施例3制备的含胆固醇结构单元和双吡啶羧酸的两亲性Tb(Ⅲ)配合物的荧光纳米纤维在ATA高效检测中的应用,其使用方法如下:
将实施例3制备的两亲性Tb(Ⅲ)配合物配制成浓度为25μmol/L(10mmol/L HEPES,pH=7.4)的两亲性Tb(Ⅲ)配合物溶液,静置半小时后,依次加入不同体积的1mmol/L ATA,配制成一系列浓度的ATA溶液:0μmol/L、2.5μmol/L、5μmol/L、7.5μmol/L、10μmol/L、12.5μmol/L、15μmol/L、20μmol/L、25μmol/L、30μmol/L、40μmol/L和50μmol/L,然后采用FLS920型单光子计数时间分辨荧光光谱仪,在320nm激发波长下,分别对以上浓度进行荧光行为测定,实验结果如图3。在图3中,可以看到随着ATA浓度的增加,荧光强度逐渐增加,当浓度增加至15μmol/L后荧光不再变化,说明与ATA的饱和浓度为15μmol/L。ATA作为Tb(Ⅲ)离子的能量给体,可协同络合纳米纤维表面的Tb(Ⅲ)离子而敏化Tb(Ⅲ)的荧光,从而实现对ATA的高效传感。最佳两亲性Tb(Ⅲ)配合物的浓度为25μmol/L。
实施例4
(1)合成6-羟甲基-2-甲酸乙酯吡啶
在流速为1.2mL/s的氮气条件下,将2,6-二甲酸乙酯吡啶和硼氢化钠加入到盛有无水乙醇的圆底烧瓶中,90℃条件下回流反应2h,然后调节pH值至5,用三氯甲烷萃取,无水硫酸钠干燥,旋蒸蒸除三氯甲烷后以甲醇与三氯甲烷的体积比为1:15的混合液为流动相、硅胶为固定相进行柱色谱分离,制备成6-羟甲基-2-甲酸乙酯吡啶。其中,2,6-二甲酸乙酯吡啶和硼氢化钠的摩尔比是1:0.5。
(2)合成6-氯甲基-2-甲酸乙酯吡啶
将6-羟甲基-2-甲酸乙酯吡啶和氯化亚砜加入到盛有无水二氯甲烷的圆底烧瓶中,冰浴条件下搅拌30min,然后室温搅拌3h后,旋蒸蒸除二氯甲烷,甲苯溶解、0.1mol/L碳酸氢钠溶液洗涤,然后水洗有机层至中性,无水硫酸钠干燥后旋蒸蒸除溶剂,得到6-氯甲基-2-甲酸乙酯吡啶。其中,6-羟甲基-2-甲酸乙酯吡啶和氯化亚砜的摩尔之比为1:1.4,6-羟甲基-2-甲酸乙酯吡啶和二氯甲烷的质量之比为1:20。
(3)合成式Ⅰ化合物
室温条件下,将胆固醇、质量分数为40%的KOH溶液、十八冠六醚和丙烯腈依次加入到盛有无水二氯甲烷的圆底烧瓶中,室温搅拌8h,蒸馏水洗涤、干燥除水,旋蒸蒸除溶剂后以乙酸乙酯与正己烷的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,得到化合式Ⅰ化合物,其中,胆固醇与KOH、十八冠六醚和丙烯腈的摩尔比为1:3.7:1:3。
(4)合成式Ⅱ化合物
将式Ⅰ化合物、六水氯化镍和二碳酸二叔丁酯加入到盛有无水甲醇的圆底烧瓶中,室温搅拌5min后,再缓慢加入硼氢化钠,室温反应12h,抽滤,旋蒸蒸除滤液后以乙酸乙酯与正己烷的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅱ化合物,其中,式Ⅰ化合物、六水氯化镍、二碳酸二叔丁酯和硼氢化钠的摩尔比为1:1:3:6,式Ⅰ化合物与无水甲醇的质量比为1:120。
(5)合成式Ⅲ化合物
将式Ⅱ化合物加入到盛有无水二氯甲烷的圆底烧瓶中,然后加入三氟乙酸,室温搅拌8h,旋蒸蒸除二氯甲烷后以甲醇与三氯甲烷的体积比为1:10的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅲ化合物,其中,式Ⅱ化合物与三氟乙酸摩尔比为1:20,三氟乙酸与二氯甲烷的体积比为1:4。
(6)合成式Ⅳ化合物
在流速为1.2mL/s的氮气条件下,向盛有乙腈的三颈烧瓶中加入式Ⅲ化合物、三乙胺,、碘化钾、碳酸钾和6-氯甲基-2-甲酸乙酯吡啶,81℃加热回流80h。然后过滤,将滤液旋蒸蒸除后以四氢呋喃与石油醚的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,用甲醇共旋,35℃真空干燥后,用正己烷重结晶、离心,得到式Ⅳ化合物,其中,式Ⅲ化合物与三乙胺、碘化钾、碳酸钾、6-氯甲基-2-甲酸乙酯吡啶的摩尔比为1:1:2.5:5:3.5,式Ⅲ化合物和乙腈的质量比为1:70。
(7)合成式Ⅴ化合物:
将式Ⅳ化合物加入到盛有无水乙醇的圆底烧瓶中,加热至80℃,恒温搅拌1h,自然冷却至室温,加入0.5mol/L的氢氧化钠水溶液,80℃条件下加热4h,然后旋蒸蒸除溶液,再逐滴滴加1mol/L的盐酸,产生白色沉淀,抽滤、真空干燥,得到式Ⅴ化合物,其中,式Ⅳ化合物与氢氧化钠的摩尔比为1:4,式Ⅳ化合物与乙醇的质量比为1:85。
(8)合成式Ⅵ化合物
将式Ⅴ化合物加入水中,再依次加入200mmol/L氢氧化钠溶液和200mmol/L HEPES缓冲溶液(pH=7.4),50℃条件下加热至固体溶解,冷却至室温后,缓慢加入10mmol/L六水氯化铽水溶液3mL,式Ⅴ化合物与六水氯化铽的摩尔比为1:1,旋蒸抽滤,所得固体即为式Ⅵ化合物,其中,式Ⅴ化合物与HEPES、氢氧化钠摩尔比为1:10:2,式Ⅴ化合物与水的质量比为1:420。
基于两亲性Tb(Ⅲ)配合物的荧光纳米纤维的制备方法:将HEPES溶于水中,通过1mmol/L的氢氧化钠水溶液调节pH值,配制成pH为7.4的HEPES缓冲溶液,然后将式化合物Ⅵ溶于10mmol/L pH值为7.4的HEPES缓冲溶液中,50℃加热3h后自然冷却至室温,得到25μmol/L的式Ⅵ化合物水溶液,即得到荧光纳米纤维。
实施例5
(1)合成6-羟甲基-2-甲酸乙酯吡啶
在流速为1mL/s的氮气条件下,将2,6-二甲酸乙酯吡啶和硼氢化钠加入到盛有无水乙醇的圆底烧瓶中,90℃条件下回流反应4h,然后调节pH值至5,用三氯甲烷萃取,无水硫酸钠干燥,旋蒸蒸除三氯甲烷后以甲醇与三氯甲烷的体积比为1:15的混合液为流动相、硅胶为固定相进行柱色谱分离,制备成6-羟甲基-2-甲酸乙酯吡啶。其中,2,6-二甲酸乙酯吡啶和硼氢化钠的摩尔比是1:0.6。
(2)合成6-氯甲基-2-甲酸乙酯吡啶
将6-羟甲基-2-甲酸乙酯吡啶和氯化亚砜加入到盛有无水二氯甲烷的圆底烧瓶中,冰浴条件下搅拌30min,然后室温搅拌2h后,旋蒸蒸除二氯甲烷,甲苯溶解、0.1mol/L碳酸氢钠溶液洗涤,然后水洗有机层至中性,无水硫酸钠干燥后旋蒸蒸除溶剂,得到6-氯甲基-2-甲酸乙酯吡啶。其中,6-羟甲基-2-甲酸乙酯吡啶和氯化亚砜的摩尔之比为1:1.7,6-羟甲基-2-甲酸乙酯吡啶和二氯甲烷的质量之比为1:50。
(3)合成式Ⅰ化合物
室温条件下,将胆固醇、质量分数为40%的KOH溶液、十八冠六醚和丙烯腈依次加入到盛有无水二氯甲烷的圆底烧瓶中,室温搅拌10h,蒸馏水洗涤、干燥除水,旋蒸蒸除溶剂后以乙酸乙酯与正己烷的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,得到化合式Ⅰ化合物,其中,胆固醇与KOH溶液、十八冠六醚和丙烯腈的摩尔比为1:3.5:1:2.5。
(4)合成式Ⅱ化合物
将式Ⅰ化合物、六水氯化镍和二碳酸二叔丁酯加入到盛有无水甲醇的圆底烧瓶中,室温搅拌5min后,再缓慢加入硼氢化钠,室温反应18h,抽滤,旋蒸蒸除溶剂后,以乙酸乙酯与正己烷的体积比为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅱ化合物,其中,式Ⅰ化合物、六水氯化镍、二碳酸二叔丁酯和硼氢化钠的摩尔比为1:1:2.5:8,式Ⅰ化合物与无水甲醇的质量比为1:150。
(5)合成式Ⅲ化合物
将式Ⅱ化合物加入到盛有无水二氯甲烷的圆底烧瓶中,然后加入三氟乙酸,室温搅拌5h,旋蒸蒸除二氯甲烷后以甲醇与三氯甲烷的体积比为1:10的混合液为流动相、硅胶为固定相进行柱色谱分离,得到式Ⅲ化合物,其中,式Ⅱ化合物与三氟乙酸摩尔比为1:30,三氟乙酸与二氯甲烷的体积比为1:4。
(6)合成式Ⅳ化合物
在流速为1mL/s的氮气条件下,将盛有乙腈的三颈烧瓶中加入式Ⅲ化合物、三乙胺、碘化钾、碳酸钾和6-氯甲基-2-甲酸乙酯吡啶,85℃加热回流70h。然后过滤,将滤液旋蒸蒸除后以四氢呋喃与石油醚的体积为1:4的混合液为流动相、硅胶为固定相进行柱色谱分离,用甲醇共旋,35℃真空干燥后,用正己烷重结晶、离心,制备成式Ⅳ化合物,其中,式Ⅲ化合物与三乙胺、碘化钾、碳酸钾、6-氯甲基-2-甲酸乙酯吡啶的摩尔比为1:1:3.5:7:2.5,式Ⅲ化合物和乙腈的质量比为1:80。
(7)合成式Ⅴ化合物
将式Ⅳ化合物加入到盛有无水乙醇的圆底烧瓶中,加热至70℃,恒温搅拌1.5h,自然冷却至室温,加入0.5mol/L的氢氧化钠水溶液,70℃条件下加热8h,然后旋蒸蒸除溶液,再逐滴滴加1mol/L的盐酸,产生白色沉淀,抽滤、真空干燥,得到式Ⅴ化合物,其中,式Ⅳ化合物与氢氧化钠的摩尔比为1:5,式Ⅳ化合物与乙醇的质量比为1:110。
(8)合成式Ⅵ化合物
将式Ⅴ化合物加入到盛有水的圆底烧瓶中,再依次加入200mmol/L氢氧化钠溶液和200mmol/L HEPES缓冲溶液(pH=7.4),50℃条件下加热至固体溶解,冷却至室温后,缓慢加入10mmol/L六水氯化铽水溶液3mL,式Ⅴ化合物与六水氯化铽的摩尔比为1:1,旋蒸抽滤,所得固体即为式Ⅵ化合物,其中,式Ⅴ化合物与HEPES、氢氧化钠摩尔比为1:10:3,式Ⅴ化合物与水的质量比为1:500。
基于两亲性Tb(Ⅲ)配合物的荧光纳米纤维的制备方法:将HEPES溶于水中,通过1mmol/L的氢氧化钠水溶液调节pH值,配制成pH为7.4的HEPES缓冲溶液,然后将式化合物Ⅵ溶于10mmol/L pH值为7.4的HEPES缓冲溶液中,52℃加热1.5h后自然冷却至室温,得到25μmol/L的式Ⅵ化合物水溶液,即得到荧光纳米纤维。
两亲性Tb(Ⅲ)配合物及其制备方法以及荧光纳米纤维的制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。












动态评分
0.0