

IPC分类号 : C08F299/02,C08J3/075,C08J3/24,C08J3/28,C08L55/00,A61L26/00,A61L24/06,A61L24/00




专利摘要
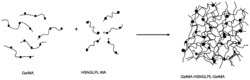
本发明公开基于线性共聚物的水凝胶及其应用,通过零价铜调节可控活性自由基聚合方法共聚聚乙二醇甲基丙烯酸酯与聚乙二醇甲醚丙烯酸酯制备侧链含有羟基的线性共聚物与丙烯酰氯制备的侧链含有乙烯基的线性聚合物为前驱体,以2,2‑二甲氧基‑苯基苯乙酮为紫外光引发剂,水为溶剂,在紫外灯照射下引发前驱体中碳碳双键进行交联为水凝胶材料。通过测试可知,线性聚合物具有一定的储存模量和粘附强度,并具有一定的溶胀性能与较慢的降解性能,以及良好的细胞相容性,在制备创伤敷料或组织粘合剂中的应用。
权利要求
1.基于线性共聚物的水凝胶,其特征在于,通过零价铜调节可控活性自由基聚合方法共聚聚乙二醇甲基丙烯酸酯与聚乙二醇甲醚丙烯酸酯制备侧链含有羟基的线性共聚物与丙烯酰氯制备的侧链含有乙烯基的线性聚合物为前驱体,以2,2-二甲氧基-苯基苯乙酮为紫外光引发剂,水为溶剂,在紫外灯照射下引发前驱体中碳碳双键进行交联为水凝胶材料,其中:将聚乙二醇甲醚丙烯酸酯、聚乙二醇甲基丙烯酸酯、CuBr
2.根据权利要求1所述的基于线性共聚物的水凝胶,其特征在于,侧链含有乙烯基的线性聚合物PDI为1.39-1.41,马克-霍温尔常数α为0.51-0.59,结构呈无规线团。
3.根据权利要求1所述的基于线性共聚物的水凝胶,其特征在于,聚乙二醇甲醚丙烯酸酯的数均分子量为480;聚乙二醇甲基丙烯酸酯的数均分子量为500。
4.根据权利要求1所述的基于线性共聚物的水凝胶,其特征在于,聚乙二醇甲醚丙烯酸酯、聚乙二醇甲基丙烯酸酯、2-溴-2-甲基丙酸乙酯、CuBr
5.根据权利要求1所述的基于线性共聚物的水凝胶,其特征在于,侧链含有羟基的线性共聚物、三乙胺、丙烯酰氯的摩尔比为1:(23—92):(21—84),选择在二氯甲烷中进行反应;反应温度为-10—-5℃,向侧链含有羟基的线性共聚物和三乙胺中滴加丙烯酰氯,滴加完毕后继续反应5-7h。
6.根据权利要求1所述的基于线性共聚物的水凝胶,其特征在于,侧链含有羟基的线性共聚物的重均分子量为10kDa—40kDa,乙烯基的含量为28—70%。
7.根据权利要求1所述的基于线性共聚物的水凝胶,其特征在于,在进行侧链含有羟基的线性共聚物的制备时,在使用缠绕铜丝的搅拌子的同时,使用铜丝加入反应体系中。
8.根据权利要求1所述的基于线性共聚物的水凝胶,其特征在于,紫外灯照射强度为0.5—2W/cm
9.根据权利要求8所述的基于线性共聚物的水凝胶,其特征在于,紫外灯照射强度为0.8—1.5W/cm
10.根据权利要求1所述的基于线性共聚物的水凝胶,其特征在于,前驱体和光引发剂的质量比为100:(1—1.2),前驱体的浓度为10—50wt%。
11.如权利要求1—10之一所述的基于线性共聚物的水凝胶在制备创伤敷料或组织粘合剂中的应用。
说明书
技术领域
本发明属于新型生物医用材料领域,主要涉及基于线性共聚物的水凝胶及其应用。
背景技术
创伤敷料是治疗急性和慢性创伤的主要方式。近几十年中,根据不同的创伤情况研究发明了多种特殊的创伤敷料。其中作为新型创伤敷料中的一种,水凝胶创伤敷料因表面光滑、生物相容性好、与不平整创面结合紧密、促进了上皮细胞生长等优点而得到了广泛使用。但目前广泛使用的合成水凝胶敷料普遍存在着伤口组织粘合能力差、力学性能与伤口组织不匹配以及生物毒性较大等问题。研究发现,通过控制合成水凝胶的所用聚合物的成分、结构、聚合度等可实现改善合成水凝胶创伤敷料的性能。线性聚合物因其结构类似于无规线团,所以其很难形成交联网络结构而制备水凝胶材料。
发明内容
本发明的目的在于克服现有技术的不足,提供基于线性共聚物的水凝胶及其应用,利用零价铜调控的可控/活性自由基聚合方法(Cu
本发明的技术目的通过下述技术方案予以实现:
线性共聚物,通过零价铜调节可控活性自由基聚合方法共聚聚乙二醇甲基丙烯酸酯与聚乙二醇甲醚丙烯酸酯制备侧链含有羟基的线性共聚物,在反应的整个过程中,聚合物分子量随单体转化率呈线性增长,且呈较窄的单峰分布,随着反应的进行,聚合物增长速率逐渐降低,反应体系中单体浓度逐渐降低,且粘度增加;再将侧链含有羟基的线性共聚物与丙烯酰氯反应,丙烯酰氯与线性聚合物侧链键接,以使线性聚合物侧链含有乙烯基。
聚乙二醇甲醚丙烯酸酯的数均分子量为480;聚乙二醇甲基丙烯酸酯的数均分子量为500。
线性共聚物的重均分子量为10kDa—40kDa,聚合物分子量随单体转化率呈线性增长,且呈较窄的单峰分布(PDI为1.39-1.41)。
线性共聚物中乙烯基的含量为28—70%。
线性共聚物的马克-霍温尔常数α为0.51-0.59,这表明线性共聚物的结构呈无规线团。
在进行侧链含有羟基的线性共聚物的制备时,反应温度为20—25摄氏度,反应时间为2—22小时。
线性共聚物的制备方法,将聚乙二醇甲醚丙烯酸酯、聚乙二醇甲基丙烯酸酯、CuBr2、2-溴-2-甲基丙酸乙酯和N,N,N′,N″,N″-五甲基二乙烯三胺均匀分散在二甲基亚砜中,并将缠绕铜丝的搅拌子置于二甲基亚砜中对反应系统进行搅拌,在除氧条件下进行反应,通过零价铜调节可控活性自由基聚合方法共聚聚乙二醇甲基丙烯酸酯与聚乙二醇甲醚丙烯酸酯制备侧链含有羟基的线性共聚物,在反应的整个过程中,聚合物分子量随单体转化率呈线性增长,且呈较窄的单峰分布,随着反应的进行,聚合物增长速率逐渐降低,反应体系中单体浓度逐渐降低,且粘度增加;再将侧链含有羟基的线性共聚物与三乙胺、丙烯酰氯反应,丙烯酰氯与线性聚合物侧链键接,以使线性聚合物侧链含有乙烯基。
聚乙二醇甲醚丙烯酸酯、聚乙二醇甲基丙烯酸酯、2-溴-2-甲基丙酸乙酯、CuBr2和N,N,N′,N″,N″-五甲基二乙烯三胺的摩尔比为(5—100):(5—25):1:0.4:0.8。
在进行侧链含有羟基的线性共聚物的制备时,反应温度为20—25摄氏度,反应时间为2—22小时。
在进行侧链含有羟基的线性共聚物的制备时,在使用缠绕铜丝的搅拌子的同时,使用铜丝加入反应体系中。
侧链含有羟基的线性共聚物、三乙胺、丙烯酰氯的摩尔比为1:(23—92):(21—84),选择在二氯甲烷中进行反应。
在进行线性共聚物的制备时,反应温度为-10—-5℃,向侧链含有羟基的线性共聚物和三乙胺中滴加丙烯酰氯,滴加完毕后继续反应5-7h。
基于线性共聚物的水凝胶,通过零价铜调节可控活性自由基聚合方法共聚聚乙二醇甲基丙烯酸酯与聚乙二醇甲醚丙烯酸酯制备侧链含有羟基的线性共聚物与丙烯酰氯制备的侧链含有乙烯基的线性聚合物为前驱体,以2,2-二甲氧基-苯基苯乙酮为紫外光引发剂,水为溶剂,在紫外灯照射下引发前驱体中碳碳双键进行交联为水凝胶材料。
紫外灯照射强度为0.5—2W/cm
前驱体和光引发剂的质量比为100:(1—1.2)。
前驱体的浓度为10—50wt%,即前驱体质量(mg)/水的体积(μL)。
本发明中采用的是新型的零价铜调控的可控/活性自由基聚合方法(Cu
本专利通过零价铜调节的可控/活性自由基聚合方法(Cu
附图说明
图1是单电子转移活性自由基聚合(SET-LRP)聚合机理示意图。
图2是单电子转移活性自由基聚合(SET-LRP)中的催化循环机理示意图。
图3是利用零价铜调控的活性自由基聚合方法制备线性共聚物的方法示意图。
图4是本发明实施例凝胶渗透色谱(GPC)测试结果图,其中a为L1,b为L2,c为L3。
图5是本发明实施例线性聚合物的核磁共振氢谱图,其中a为改性前,b为改性后。
图6是本发明实施例不同分子量的线性聚合物的马克-霍温克公式常数α的示意图。
图7是本发明实施例共聚物水凝胶的流变测试曲线图,其中a为线性水凝胶的震荡-时间光交联的流变测试图,b为线性水凝胶的震荡-频率模式的流变测试图。
图8是本发明实施例浓度为50%的不同分子量的线性水凝胶材料的溶胀性能测试曲线图。
图9是本发明实施例浓度为50%的不同分子量的线性水凝胶材料的降解性能测试曲线图。
具体实施方式
下面结合具体实施例进一步说明本发明的技术方案。使用原料和仪器如下表所示:
实验原料
实验仪器
利用零价铜调控的活性自由基聚合方法制备线性共聚物的方法如下:
(1)配置5mg/mL的CuBr2溶液:称取100mg的CuBr2粉末,放入20mL的一次性透明玻璃瓶中,加入20mL的二甲基亚砜(DMSO)。将其置于超声仪中约5min,待CuBr2完全溶解后取出备用。
(2)分子量为10kDa的线性共聚物的配方为:依次准确称量聚乙二醇甲醚丙烯酸酯(PEGA480,6mmol,2.88g),聚乙二醇甲基丙烯酸酯(PEGMA500,6mmol,3.00g),DMSO(33.19mL),CuBr2溶液(0.48mmol,21.44mL),2-溴-2-甲基丙酸乙酯(EBriB,1.2mmol,178.00μL),N,N,N′,N″,N″-五甲基二乙烯三胺(PMDETA,0.96mmol,200.44μL),加入100mL的两口圆底烧瓶中。其中PEGA480:PEGMA500:EBriB:CuBr2:PMDETA=5:5:1:0.4:0.8。
分子量为20kDa的线性共聚物的配方为:依次准确称量PEGA480(7.2mmol,3.46g),PEGMA500(4.8mmol,2.40g),DMSO(46.07mL),CuBr2溶液(0.19mmol,8.58mL),EBriB(0.48mmol,71.20μL),PMDETA(0.38mmol,80.18μL),加入100mL的两口圆底烧瓶中。其中PEGA480:PEGMA500:EBriB:CuBr2:PMDETA=15:10:1:0.4:0.8(摩尔比)。
分子量为40kDa的线性共聚物的配方为:依次准确称量PEGA480(9.6mmol,4.61g),PEGMA500(2.4mmol,1.20g),DMSO(52.97mL),CuBr2溶液(0.04mmol,1.72mL),EBriB(0.10mmol,14.24μL),PMDETA(0.08mmol,16.04μL),加入100mL的两口圆底烧瓶中。其中PEGA480:PEGMA500:EBriB:CuBr2:PMDETA=96:24:1:0.4:0.8。
先后分别用清洗干净的橡胶塞和封口膜密封,通氩气30min以除氧。与此同时,将直径为1mm,长5cm的铜丝,均匀缠绕并固定于棒状搅拌子上。先用浓度为32%的盐酸(质量百分数32wt%的氯化氢水溶液)浸泡10min,然后再依次用丙酮,超纯水及丙酮清洗,并干燥。
(3)待除氧30min后,迅速打开圆底烧瓶的其中一个瓶口并将处理干净的铜丝加入反应体系中,再次密封,继续除氧2min左右。除氧结束后,将两口圆底烧瓶置于25℃,700r/min的油浴加热器中,开始反应并计时。
(4)在间隔相同的时间时,先通氩气,然后用清洗干净的5mL注射器置入液面以下,通过正压使样品流到注射器中。待样品量达2mL时,将注射器取出然后停止通氩气。将取得的样品置于20mL的一次性玻璃瓶中,并标记。用移液枪从中取100μL样品并用DMF(二甲基甲酰胺)稀释至1mL,充分混合均匀。选择小型氧化铝柱,并用DMF润湿,然后过滤稀释后的聚合物样品,除去其中的Cu。此时可观察到样品由浅蓝色变透明。之后用直径为0.4mm的滤头过滤样品,最后将样品放置于GPC测试小瓶中,并标记测试。
(5)通过GPC监测反应的进行,待分子量达到目的分子量时,将两口圆底烧瓶从油浴加热器中取出,并打开密封的瓶口,使其充分与空气接触,用磁棒将缠有铜丝的磁子取出。在清洗干净的1000mL的大烧杯中加入5-7倍反应原液体积的乙醚,设定转速为600r/min。在高速转动的条件下,将反应原液通过分液漏斗逐滴滴加至乙醚中并用锡纸将烧杯封口。待滴加结束后,继续搅拌30min左右,室温下静置5-7h。待混合液静置分层且上层液较为清澈透明时,将上层清液倒出。继续在高速搅拌的状态下加入3-5倍下层液体积的乙醚,在锡纸封口的状态下搅拌约30min,再次静置分层。如此反复两次后,下层的聚合物粘度逐渐增加并粘附在烧杯底部。
(6)选择中型的氧化铝柱,先依次加入少量的棉花及沙子,使其铺平。然后加入约3/5氧化铝柱高度的氧化铝粉末,制备氧化铝过滤柱。使用前用丙酮将其润湿。用少量丙酮稀释沉淀后聚集在烧杯底部的聚合物,充分溶解后沿柱壁倒入氧化铝过滤柱中,用称量好质量的一次性玻璃瓶收集过滤后的清澈聚合物溶液。
(7)将过滤后收集到的所有产物都用锡纸封口并均匀的扎上小孔,然后放置于真空干燥箱,除去溶剂,得到透明的纯聚合物。然后称重,计算产率。
制备得到的线性共聚物侧链中含有羟基(-OH),通过与丙烯酰氯可制备侧链含有乙烯基的线性共聚物。类似的,分子量不同的线性共聚物-OH含量不同,故所需添加的试剂的量不同。具体的制备过程如下:
(1)分子量为10kDa的线性共聚物:依次准确称量线性共聚物(0.08mmol,0.80g),三乙胺(TEA,1.84mmol,256.46μL),二氯甲烷(20mL)加入50mL的两口圆底烧瓶中。丙烯酰氯(1.68mmol,136.50μL)溶解于5mL二氯甲烷中,并逐滴滴加至反应体系中。其中聚合物:TEA:丙烯酰氯=1:23:21(摩尔比)。
分子量为20kDa的线性共聚物:依次准确称量线性共聚物(0.04mmol,0.80g),TEA(1.84mmol,256.46μL),二氯甲烷(20mL)加入50mL的两口圆底烧瓶中。丙烯酰氯(1.68mmol,136.50μL)溶解于5mL二氯甲烷中,并逐滴滴加至反应体系中。其中聚合物:TEA:丙烯酰氯=1:46:42。
分子量为40kDa的线性共聚物:依次准确称量线性共聚物(0.05mmol,2.00g),TEA(4.60mmol,641.15μL),二氯甲烷(20mL)加入50mL的两口圆底烧瓶中。丙烯酰氯(4.20mmol,341.241μL)溶解于5mL二氯甲烷中,并逐滴滴加至反应体系中。其中聚合物:TEA:丙烯酰氯=1:92:84。
滴加丙烯酰氯的反应过程中,使反应稳定在-10℃,700r/min的条件下。待滴加完成后,在-5℃,700r/min的条件下继续反应5-7h。
(2)待反应结束后,将反应原液用放有棉花并用二氯甲烷润湿的漏斗过滤掉反应中的无机盐沉淀,并用旋转蒸发仪除去反应原液中的二氯甲烷。然后将得到的产物在超纯水中用规格为1000Da的透析袋进行透析,每天换水3-5次,透析2-3天,除去原液中剩余的少量无机盐。
(3)待透析结束后,将聚合物溶液放置于称量好质量的20mL一次性玻璃瓶中,冷冻干燥,得到最终的聚合物,称重并计算产率。
如图4的凝胶渗透色谱(GPC)测试结果可知,在反应的整个过程中,聚合物分子量随单体转化率呈线性增长,且呈较窄的单峰分布(PDI为1.39-1.41)。随着反应的进行,聚合物增长速率逐渐降低,这是由于随着反应的进行,反应体系中单体浓度逐渐降低,且粘度增加,单体与增长的聚合物的反应更难。这是典型的线性聚合物的聚合特征。由图4(b)可知三种聚合物均为分子量分布较窄的线性共聚物。本专利中,分别将分子量为10kDa、20kDa、40kDa的线性共聚物命名为L1、L2、L3。
通过对不同分子量的线性共聚物poly(PEGMA500-co-PEGA480)L进行核磁共振氢谱测试,可知聚合物与丙烯酰氯反应前后,聚合物中的羟基(-OH)被丙烯酸酯基(-O-CO-CH=CH2)所取代,即不同分子量的线性共聚物中存在着数量不等的乙烯基官能团。聚合物中乙烯基的含量(即支化度)可通过公式(1)计算,a和h分别为峰值(即峰面积大小)。
如图5所示,线性共聚物与丙烯酰氯反应前,在化学位移δ=4.5-4.6处有吸收峰,此为羟基吸收峰。反应后,在化学位移δ=5.7-6.5处有吸收峰,此为双键吸收峰。由上述公式计算可知,线性共聚物中乙烯基含量因其分子量不同而不同。分子量为10kDa、20kDa及40kDa的线性共聚物中乙烯基的含量分别为67.41%,53.71%及28.38%。
聚合物在溶剂中的构象与其马克-霍温克公式常数α有关。当α≤0.5时,聚合物呈现较为致密的结构。当0.5≤α≤0.8时,聚合物呈现无规线团构象;而聚合物线团越为伸展,α越接近0.8;聚合物呈刚性线团状时,1≤α。聚合物的α值可通过GPC三种检测器(差折光检测RI,粘度检测器VS和激光散射检测器LS)的联用来测得。测试结果如图6所示,结果表明线性共聚物的马克-霍温尔常数α为0.51-0.59(表1),这表明线性共聚物的结构呈无规线团。
表1不同分子量的单链超内环化聚合物的反应结果
由
分别以分子量不同的线性共聚物为前驱体,2,2-二甲氧基-苯基苯乙酮(Irgacure2959)为紫外光引发剂,水为溶剂,采用光引发自由基聚合反应制备了一系列不同浓度的水凝胶材料。具体制备方法如下:依照表2,将不同分子量的线性共聚物配置为不同浓度的聚合物水溶液,置于一次性玻璃瓶中并充分溶解。由于Irgacure 2959在纯净水中的溶解性很差,故将其溶解于丙酮中,且配为质量浓度为5%的Irgacure 2959/丙酮溶液,在光交联前用移液枪取相应体积的溶液加入到聚合物水溶液中,迅速用涡旋仪震荡混匀。取适量体积的无色透明溶液置于相应容器内,放于UV灯下,在光照条件下固化成水凝胶。
表2制备水凝胶材料的投料比
聚合物的流变性能及紫外引发固化成胶的研究可通过由压力控制的平板型(d=8mm)AR2000流变仪来实现。在光交联流变性能的研究中,采用的是波长为320-390nm,光强度为100mW/cm
在震荡-时间测试模式下,聚合物水溶液的储存模量G'和损耗模量G"在前1min内无明显变化。紫外光照后,聚合物的G'在1min后开始逐渐增加且大于G",在20秒内G'与G"出现了交点,表明了水凝胶材料的形成。从图7(a)可知,交联后的水凝胶材料的模量比未交联聚合物的模量高了1-2个数量级,且达到恒定值而不随时间变化。此外,线性共聚物的G'随分子量的增加而降低(L1:5.6kPa,L2:2.4kPa,L3:0.36kPa),这是由于随着聚合物分子量的增加,其乙烯基含量较少,交联密度降低,从而导致G'的下降。
在震荡-频率测试模式下,对水凝胶材料的稳定性进一步表征。如图7(b)所示,在0.01Hz到256Hz的测试频率条件下,线性水凝胶的G'在0.01Hz-50Hz频率范围内一直保持稳定。
为了表征水凝胶材料的粘附性能,可通过Lap-shear,Pull-off,Burst测试分别从横向粘附、纵向粘附及耐冲击程度来进行力学粘附性能的表征。
Lap-shear测试
进行Lap-shear测试前需要准备测试样品。具体制备过程如下:
1)配置聚合物混合溶液:将不同结构、不同分子量的纯聚合物分别配置为不同浓度(10%,20%,30%,50%w/v)的聚合物水溶液;然后将紫外光引发剂Irgacure 2959配置成浓度为5%的丙酮溶液。按照聚合物与光引发剂的质量比为100:1的比例混合均匀。
2)粘附试样的制备:将预处理过的猪皮裁剪为长40mm,宽25mm,厚1mm的形状,用Superglue将其脂肪侧粘在长75mm,宽25mm,厚1mm的玻璃片上。然后用移液枪取200μL的聚合物混合溶液使其均匀的平铺在猪皮表皮上。再取同样大小的玻璃片使其轻轻覆盖在聚合物混合溶液上。在预设光强和时间条件下(强度:0.8,1.2,1.7W/cm
3)Lap-shear测试:将制备好的测试样品,垂直方向上平行的置于测试机上。在2mm/min的恒定速度下进行拉伸,直至断裂。粘附强度为断裂前的最大值,每组测试重复3次。
Pull-off测试
进行Pull-off测试前需要准备测试样品。具体制备过程如下:
1)配置聚合物混合溶液:将不同结构、不同分子量的纯聚合物分别配置为不同浓度(10%,20%,30%,50%w/v)的聚合物水溶液;然后将紫外光引发剂Irgacure 2959配置成浓度为5%的丙酮溶液。按照聚合物与光引发剂的质量比为100:1的比例混合均匀。
2)粘附试样的制备:将预处理过的猪皮裁剪为直径25mm,厚1mm的圆片形,用Superglue将其脂肪侧粘在直径为25mm的铝片上。然后用移液枪取100μL的聚合物混合溶液使其均匀的平铺在猪皮表皮上。再用长75mm,宽25mm,厚1mm的玻璃片轻轻覆盖在聚合物混合溶液上。在预设光强和时间条件下(强度:1.7W/cm
3)Pull-off测试:将制备好的测试样品,水平方向上平行的置于测试机上。在2mm/min的恒定速度下进行拉伸,直至断裂。粘附强度为断裂前的最大值,每组测试重复3次。
Burst测试
进行Burst测试前需要准备测试样品。具体制备过程如下:
1)配置聚合物混合溶液:将不同结构、不同分子量的纯聚合物分别配置为不同浓度(10%,20%,30%,50%w/v)的聚合物水溶液;然后将紫外光引发剂Irgacure 2959配置成浓度为5%的丙酮溶液。按照聚合物与光引发剂的质量比为100:1的比例混合均匀。
2)粘附试样的制备:将预处理过的猪皮裁剪为直径30mm,厚1mm的圆片形,用Superglue将其脂肪面固定在Burst测试泵的表面,并在猪皮的表面扎一个小孔,与泵上小孔位置平行,大小一致。打开水龙头,调节流速使得水能以水柱的形式喷出并记录此时的初始压力P0。关闭水龙头并除去猪皮表面的水。然后用移液枪取200μL的聚合物混合溶液使其均匀的平铺在猪皮表皮上。在预设光强和时间条件下(强度:1.7W/cm
3)Burst测试:打开水龙头,在预设流速条件下进行测试,直至水凝胶破裂,水柱喷出,记录最大压力值Pt。则最大粘附强度为P=Pt-P0,每组测试重复3次。
如表3所示,在最优化光交联条件下(在紫外灯照射强度为0.5—2W/cm
表3线性水凝胶材料的粘附性能测试
聚合物水凝胶是由水和聚合物网络结构构成的,它可以吸收一定量的水分并溶胀为溶胀水凝胶。平衡状态的溶胀率可视为表征聚合物交联度的一个直接参数,因此可通过溶胀实验来表征聚合物的结构特征。溶胀率会随着交联度的不同而变化。
水凝胶溶胀性能的测试可用称重法进行测定。具体过程如下:
1)水凝胶的制备:将不同结构、不同分子量的聚合物配置为浓度为30%的聚合物溶液,然后加入浓度为5%的光引发剂Irgacure 2959溶液,质量比为100:1。取50μL的混合物溶液置于已称重的玻璃片上,在强度为1.7W/cm
2)称重:定期的从PBS缓冲液中取出溶胀的水凝胶,轻轻擦去表面多余水分并称重,记为Wt。然后将水凝胶重新放回PBS缓冲液中。水凝胶的溶胀率(SR)可通过公式(3-1)计算:
SR=(Wt-W0)/W0×100%(公式3-1)
每种水凝胶取四个样品进行测试,求平均值,记为最终的溶胀率SR。
为了在生理条件下评价水凝胶的溶胀性能,测试时将制备好的水凝胶浸泡在PBS缓冲液的24孔板中,并置于37℃的摇床中。溶胀率可通过一定时间内水凝胶材料重量的变化来计算。溶胀测试结果如图8所示,线性水凝胶在初期阶段溶胀较快并在10天左右时候达到平衡状态,此时的体积为原来的1.8倍。当聚合物分子量分别为10kDa、20kDa、40kDa时,线性水凝胶平衡时的溶胀率分别为65.82%、76.28%、83.37%。这是由于线性共聚物的无规线团结构使其很难形成致密的聚合物网络结构,因此溶剂分子很容易进入分子间且使得分子链得到扩展,且水凝胶材料的乙烯基含量随着分子量的增加而降低,故其交联密度下降。溶剂分子更易进入聚合物网络中,使得分子链扩张,进而引起聚合物体积的增大。
一般来说,对于大多数组织工程所应用的理想生物材料应具有可调节、较稳定的降解性能。聚合物在氧化反应、辐射、热分解或水解作用下,其主链或侧链发生断裂而引起聚合物的降解。其中,聚合物因水解作用而发生的降解可认为是聚合物中的主链,低聚物或单体间在水解作用下发生了化学键的断裂。
水凝胶降解性能的测试可用称重法进行测定。具体过程如下:
1)水凝胶的制备:将不同结构、不同分子量的聚合物配置为浓度为30%的聚合物溶液,然后加入浓度为5%的光引发剂Irgacure 2959溶液,质量比为100:1。取50μL的混合物溶液置于已称重的玻璃片上,在强度为1.7W/cm
2)称重:定期的从PBS缓冲液中取出水凝胶,经冷冻干燥处理达恒重时,记录重量Wt。水凝胶降解后残余质量百分数率可通过公式(3-2)计算:
Mass loss=(W0-Wt)/Wt×100%(公式3-2)
每种水凝胶取四个样品进行测试,求平均值,记为最终的残余质量百分数。
因线性聚合物中存在酯基官能团,当浸泡在PBS缓冲液中后会因水解作用而发生降解,实验结果如图9所示。线性水凝胶没有表现出较大的降解行为,这是由于其拥有类似于非降解性PEG水凝胶材料的较长的碳主链,仅有侧链的酯基在水解作用下发生了降解,最终聚合物降解为较长的碳链聚合物和较短的PEG链单元。当聚合物分子量不同时,其降解行为也有所变化。当聚合物分子量分别为L1、L2、L3时,聚合物降解后剩余聚合物质量分数为72.36%,80.68%,87.38%。这种变化趋势的主要原因随着聚合物分子量的增加,聚合物形成的结构相对于分子量较小的聚合物来说更为致密,水解作用相对减弱,降解能力降低,剩余聚合物的质量分数相对增加。综上所述,具有较慢降解行为的线性水凝胶可能适合用于需长期愈合的创伤敷料或组织粘合剂。
为了表征水凝胶材料的生物相容性,可通过纤维母细胞(Fibroblast)代谢活动在标准的 条件下来测试。在标准细胞培养条件下培养小鼠3T3成纤维细胞24h后,放入光交联制备的水凝胶材料。继续培养24小时后,取出水凝胶材料并用 方法来测试其细胞毒性。测试结果如表4所示,水凝胶材料的细胞毒性随聚合物分子量的增加而降低。如分子量为L1、L2、L3的线性水凝胶的细胞存活率分别为75.6%,76.55%,79.36%。这是由于随着分子量的增加,聚合物中乙烯基含量降低,故其细胞毒性降低。
表4浓度为50%的不同分子量的线性水凝胶材料的细胞毒性测试
本专利通过零价铜调控的可控/活性自由基聚合方法(Cu
根据本发明内容进行工艺参数的调整均可制备本发明的共聚物和水凝胶,且表现出与实施例基本一致的性能。以上对本发明做了示例性的描述,应该说明的是,在不脱离本发明的核心的情况下,任何简单的变形、修改或者其他本领域技术人员能够不花费创造性劳动的等同替换均落入本发明的保护范围。
基于线性共聚物的水凝胶及其应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。















动态评分
0.0