

IPC分类号 : A61L24/00,A61L24/08,A61L24/10,D01D5/00,D01F9/00,D01F4/00








专利摘要
本发明提供一种微纤维态止血材料及其制备方法和止血制品。所述微纤维态止血材料包括微纤维,所述微纤维源自于交联的纳米纤维材料,所述微纤维的直径为1μm~500μm,长度为0.5mm~10mm;所述微纤维具有由多根纳米短纤维相互搭接形成的交错结构;所述纳米短纤维的直径在1nm~1000nm之间,长度在10mm以下。本发明的微纤维态止血材料高度蓬松,具有超高的比表面积,优异的组织粘附性能,不仅促进了其与组织的相互融合过程,同时还进一步提高了材料的吸水能力与组织粘附能力,而且在大量吸液的同时能很好地维持纤维形态。
权利要求
1.一种微纤维态止血材料,其特征在于,所述微纤维态止血材料包括微纤维,
所述微纤维源自于交联的纳米纤维材料,所述微纤维的直径为1μm~500μm,长度为0.5mm~10mm;
所述微纤维具有由多根纳米短纤维相互搭接形成的交错结构;
所述纳米短纤维的直径在1nm~1000nm之间,长度在10mm以下。
2.根据权利要求1所述的微纤维态止血材料,其特征在于,所述微纤维态止血材料的堆密度小于0.03g/cm3,优选为0.01g/cm3~0.025g/cm3。
3.根据权利要求1或2所述的微纤维态止血材料,其特征在于,所述微纤维态止血材料的比表面积为4m2/g~50m2/g,优选为6m2/g~30m2/g。
4.根据权利要求1-3任一项所述的微纤维态止血材料,其特征在于,所述微纤维态止血材料具有大于1500%,优选在2000%~2500%之间的吸水率。
5.根据权利要求1-4任一项所述的微纤维态止血材料,其特征在于,所述微纤维的长度与直径的比值在5~8000的范围内。
6.根据权利要求1-5任一项所述的微纤维态止血材料,其特征在于,所述纳米纤维材料源自具有生物相容性以及可生物体降解吸收的聚合物材料,所述纳米纤维材料由纤维丝交织而成;优选地,所述纳米纤维材料为纤维团、纤维束或纤维膜。
7.根据权利要求1-6任一项所述的微纤维态止血材料,其特征在于,所述交联为在交联剂存在下的交联,优选地,所述交联剂与所述纳米纤维材料的质量比为0.1:1~3:1,优选0.5:1~2:1。
8.根据权利要求1-7任一项所述的微纤维态止血材料,其特征在于,所述微纤维态止血材料还包含有药物;优选地,所述药物包括凝血因子、生长因子中的一种或两种的组合。
9.一种根据权利要求1-8任一项所述的微纤维态止血材料的制备方法,其特征在于,包括以下步骤:
静电纺丝步骤:通过静电纺丝制备所述纳米纤维材料;
交联步骤:在交联剂的存在下对所述纳米纤维材料进行交联处理,得到交联的纳米纤维材料;
剪切步骤:对所述交联的纳米纤维材料进行剪切处理。
10.根据权利要求9所述的微纤维态止血材料的制备方法,其特征在于,所述交联剂包括:碳化二亚胺、N-羟基琥珀酰亚胺、京尼平或醛类化合物中的一种或两种以上的组合,
优选地,所述交联剂包括碳化二亚胺和N-羟基琥珀酰亚胺,更优选地,所述碳化二亚胺与N-羟基琥珀酰亚胺的质量比为1~4:1。
11.根据权利要求9或10所述的微纤维态止血材料的制备方法,其特征在于,所述剪切处理包括预剪切步骤,
所述预剪切步骤为在5000~10000rpm/min的转速下对所述交联的纳米纤维材料进行初步剪切处理,得到预剪切物。
12.根据权利要求11所述的微纤维态止血材料的制备方法,其特征在于,所述剪切处理还包括高速剪切步骤,
所述高速剪切步骤为在20000~40000rpm/min的转速下,对所述预剪切物进行高速剪切处理;
优选地,所述高速剪切处理的时间为1~10min。
13.根据权利要求9-12任一项所述的微纤维态止血材料的制备方法,其特征在于,所述剪切处理是在通入流动气体的状态下进行的。
14.根据权利要求9-13任一项所述的微纤维态止血材料的制备方法,其特征在于,所述交联步骤与剪切步骤之间还包括:洗脱步骤和/或冷冻干燥步骤。
15.根据权利要求14所述的微纤维态止血材料的制备方法,其特征在于,所述洗脱步骤包括:
在0~20℃的低温下利用洗脱剂,通过浓度梯度法洗脱所述交联的纳米纤维材料,以去除未反应的交联剂。
16.一种止血制品,其特征在于,包括根据权利要求1-8任一项所述的微纤维态止血材料,或者根据权利要求9-15任一项所述的微纤维态止血材料的制备方法得到的微纤维态止血材料。
说明书
技术领域
本发明涉及一种微纤维态止血材料及其制备方法和应用,属于生物医用材料领域。
背景技术
生物医用材料是近三十年来发展起来的一类高新技术材料,其中止血材料也随着交通意外、严重的烧烫伤以及重大灾害等事故的增多逐渐引起医学界的关注。随着现代科学技术的高速发展,止血材料的研究取得了非常快的进展,各种新型止血材料不断出现,性能也得到了很大提升。目前,常用的局部止血材料有纤维蛋白胶、凝血酶粉、明胶海绵、胶原蛋白海绵、壳聚糖海绵、氧化纤维素、微纤维胶原、海藻酸纤维、沸石、氰基丙烯酸酯、植物多糖粉等。止血效果确切、使用方便、生物相容性好、能控制降解速率的生物医用止血材料成为人们关注和研究的主要对象。
常用的止血材料的形态包括多种形式,有粉状:如凝血酶冻干粉、植物多糖粉、沸石粉、微纤维胶原粉;有溶液型,如氰基丙烯酸酯、壳聚糖溶液;有液体型但在创面能形成凝胶或胶体,如纤维蛋白胶、戊二醛-白蛋白Bioglue;膜状,如壳聚糖膜、聚乳酸膜;还有海绵状,如胶原蛋白海绵、明胶海绵、微纤维胶原海绵、纤维蛋白贴等。各种形态的止血材料各有其优点,也有各自的应用优势,主要根据创面类型和临床治疗方式进行选择。
其中,粉状的止血材料因其操作简便、不受创面部位的限制在手术止血领域具有广泛的应用。现有技术中,粉状的止血材料通过超声波法、湿热法处理、微波法、机械法或酶穿孔等技术来实现材料表面的多微孔化,提升材料的比表面积和亲水性能,在伤口表面起分子筛的作用,通过吸附血液中的水分来提升凝血因子的浓度,加速凝血机制的发生,从而实现止血作用。但是目前的止血粉主要为微球状或颗粒状,存在粘附性差,相对密实,不能采用镊子使用,制备工艺复杂等问题,而且需要提前准备,浪费不少抢救止血的时间。
发明内容
发明要解决的问题
本发明的目的是提供一种微纤维态止血材料,其具有高蓬松度、优异的粘附性能和显著的止血效果。
进一步地,本发明的微纤维态止血材料还具有高比表面积、高吸水率以及良好的生物相容性,并且可以快速地被生物体降解吸收。
用于解决问题的方案
本发提供一种微纤维态止血材料,其中,所述微纤维态止血材料包括微纤维;
所述微纤维源自于交联的纳米纤维材料,所述微纤维的直径为1μm~500μm,长度为0.5mm~10mm;
所述微纤维具有由多根纳米短纤维相互搭接形成的交错结构;
所述纳米短纤维的直径在1nm~1000nm之间,长度在10mm以下。
根据本发明的微纤维态止血材料,其中,所述微纤维态止血材料的堆密度小于0.03g/cm3,优选为0.01g/cm3~0.025g/cm3。
根据本发明的微纤维态止血材料,其中,所述微纤维态止血材料的比表面积为4m2/g~50m2/g,优选为6m2/g~30m2/g。
根据本发明的微纤维态止血材料,其中,所述微纤维态止血材料具有大于1500%,优选在2000%~2500%之间的吸水率。
根据本发明的微纤维态止血材料,其中,所述微纤维的长度与直径的比值在5-8000的范围内。
根据本发明的微纤维态止血材料,其中,所述纳米纤维材料源自具有生物相容性以及可生物体降解吸收的聚合物材料,所述纳米纤维材料由纤维丝交织而成;优选地,所述纳米纤维材料为纤维团、纤维束或纤维膜。
根据本发明的微纤维态止血材料,其中,所述交联为在交联剂存在下的交联,优选地,所述交联剂与所述纳米纤维材料的质量比为0.1:1~3:1,优选0.5:1~2:1。
根据本发明的微纤维态止血材料,其中,所述微纤维态止血材料还包含有药物;优选地,所述药物包括凝血因子、生长因子中的一种或两种的组合。
本发明还提供一种根据本发明的微纤维态止血材料的制备方法,包括以下步骤:
静电纺丝步骤:通过静电纺丝制备所述纳米纤维材料;
交联步骤:在交联剂的存在下对所述纳米纤维材料进行交联处理,得到交联的纳米纤维材料;
剪切步骤:对所述交联的纳米纤维材料进行剪切处理。
根据本发明的微纤维态止血材料的制备方法,其中,所述交联剂包括:碳化二亚胺、N-羟基琥珀酰亚胺、京尼平或醛类化合物中的一种或两种以上的组合;
优选地,所述交联剂包括碳化二亚胺和N-羟基琥珀酰亚胺,更优选地,所述碳化二亚胺与N-羟基琥珀酰亚胺的质量比为1~4:1。
根据本发明的微纤维态止血材料的制备方法,其中,所述剪切处理包括预剪切步骤;
所述预剪切步骤为在5000~10000rpm/min的转速下对所述交联的纳米纤维材料进行初步剪切处理,得到预剪切物。
根据本发明的微纤维态止血材料的制备方法,其中,所述剪切处理还包括高速剪切步骤;
所述高速剪切步骤为在20000~40000rpm/min的转速下,对所述预剪切物进行高速剪切处理;
优选地,所述高速剪切处理的时间为1~10min。
根据本发明的微纤维态止血材料的制备方法,其中,所述剪切处理是在通入流动气体的状态下进行的。
根据本发明的微纤维态止血材料的制备方法,其中,所述交联步骤与剪切步骤之间还包括:洗脱步骤和/或冷冻干燥步骤。
根据本发明的微纤维态止血材料的制备方法,其中,所述洗脱步骤包括:
在0-20℃的低温下利用洗脱剂,通过浓度梯度法洗脱所述交联的纳米纤维材料,以去除未反应的交联剂。
本发明还提供一种止血制品,其包括根据本发明的微纤维态止血材料,或者根据本发明的微纤维态止血材料的制备方法得到的微纤维态止血材料。
发明的效果
本发明的微纤维态止血材料可以高度蓬松,具有优异的组织粘附性能和显著的止血效果,不仅促进了其与组织的相互融合过程,同时还进一步提高了材料的吸水能力与组织粘附能力,而且在大量吸液的同时能很好地维持纤维形态。
进一步地,该微纤维态止血材料还具有超高的比表面积,良好的生物性能和显著的止血效果,不仅能够快速地被生物体降解吸收,而且临床使用方便,可用于创伤救治和临床手术中的止血。
附图说明
图1是本发明实施例2所述微纤维态止血材料的SEM电镜示意图。
图2是本发明实施例2单根微纤维的SEM电镜示意图。
图3是本发明实施例2所述微纤维态止血材料与市售产品1的止血有效性对比分析图。
图4是本发明实施例2所述微纤维态止血材料与市售产品1在动物实验中止血情况以及体内降解的示意图。
具体实施方式
以下将参考附图详细说明本公开的各种示例性实施例、特征和方面。在这里专用的词“示例性”意为“用作例子、实施例或说明性”。这里作为“示例性”所说明的任何实施例不必解释为优于或好于其它实施例。
另外,为了更好地说明本公开,在下文的具体实施方式中给出了众多的具体细节。本领域技术人员应当理解,没有某些具体细节,本公开同样可以实施。在另外一些实例中,对于本领域技术人员熟知的方法、手段、器材和步骤未作详细描述,以便于凸显本公开的主旨。
第一实施方式
本发明的第一实施方式提供一种微纤维态止血材料。所述微纤维态止血材料包括微纤维,所述微纤维具有由多根纳米短纤维相互搭接形成的交错结构;所述微纤维源自于交联的纳米纤维材料。
其中,交联的纳米纤维材料可以通过对纳米纤维材料进行交联改性处理得到。
微纤维可以通过对交联的纳米纤维材料进行剪切得到。经剪切同时可以形成纳米短纤维,使得微纤维具有由多根纳米短纤维相互搭接而形成的交错结构。
<纳米纤维材料>
本发明的纳米纤维材料可以源自具有生物相容性以及可生物体降解吸收的聚合物材料,更优选为亲水性好的聚合物材料。举例而言,聚合物材料可以包括胶原(Collagen)、壳聚糖(Chitosan)、透明质酸(HA)、海藻酸盐、纤维素及其衍生物中的一种或两种以上的组合。
本发明的纳米纤维材料可以由纤维丝交织而成。优选为使用静电纺丝方法制备的纳米纤维材料。所述纳米纤维材料可以为纤维团、纤维束或纤维膜,优选为纤维膜。
静电纺丝的原理是在静电纺丝过程中,对聚合物液体施加高电压,使电荷引入液体。当液体中的电荷聚集到一定量的时候,液体会在喷头形成泰勒锥,在外加电场力的作用下克服表面张力形成液体射流,然后射流在静电斥力、库伦力(Coulomb)和表面张力的共同作用下,聚合物射流沿不规则螺旋状轨迹运动。射流在极短时间内被牵引拉伸,随着溶剂挥发或者热量散失,聚合物射流固化形成微米/纳米纤维。静电纺丝过程中,很多参数会对最终静电纺丝纤维产生影响,通过控制过程参数,可以制备获得不同尺寸、形态和不同结构的微米/纳米纤维。
本发明的静电纺丝过程中,工艺参数会对静电纺丝得到的纳米纤维材料产生影响,通过控制工艺参数,可以制备获得不同尺寸、形态和不同结构的纳米纤维材料。本发明对于静电纺丝的方式没有特别的要求,可以是本领域中常用的静电纺丝方式。具体而言,本发明将聚合物材料溶于合适的溶剂中,制备聚合物材料的纺丝原液;然后采用静电纺丝将纺丝原液纺制成由纤维丝交织而成的纳米纤维材料。优选地,所述纳米纤维材料具有多孔结构。
<交联的纳米纤维材料>
本发明的交联的纳米纤维材料可以通过对纳米纤维材料进行交联改性处理得到。具体而言,交联改性是在交联剂存在下的交联改性,从而获得交联度合适,并且均匀一致的交联产品。
在本发明中,进行交联改性的目的是为了使微纤维态止血材料在大量吸液的同时,能够很好的维持纤维形态,不会很快就被所吸收的体液溶解或冲散。另外,由于过高的交联度会影响吸水率、柔软性等,为了获得更合适的交联度,本发明中交联剂与纳米纤维材料的质量比为0.1:1~3:1,优选0.5:1~2:1。
<纳米短纤维>
本发明的纳米短纤维的直径为1nm~1000nm之间,长度一般在10mm以下,可以在8mm以下,可以在5mm以下。一般而言,狭义上的纳米纤维为直径在1~100nm范围内的纳米纤维,广义上的纳米纤维还包括纳米复合纤维,即零维或一维纳米材料与常规纤维复合而成传统纤维。对于聚合物纳米料纤维而言,由于其在1000nm的尺度上就已经产生了许多特殊性能,如巨大的表面积、易表面功能化和优越的机械性能等,因此本发明中纳米纤维是指直径在1~1000nm范围内的纳米纤维。
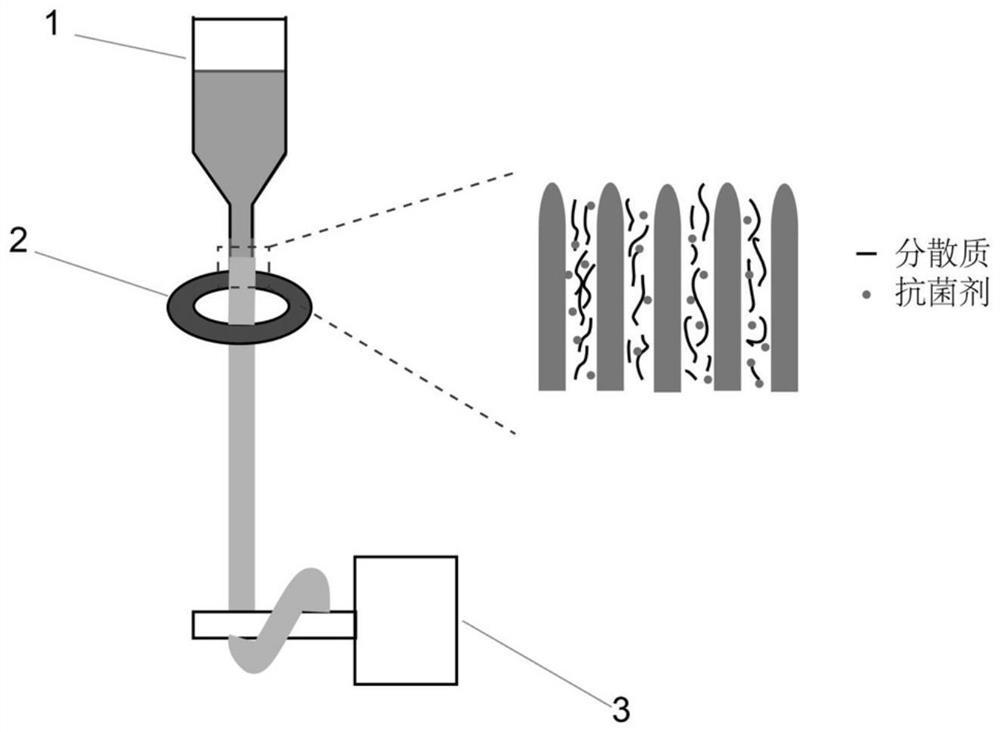
<微纤维>
本发明的微纤维可以通过对交联的纳米纤维材料进行剪切得到。如图2所示,经剪切同时形成纳米短纤维,使得微纤维具有由多根纳米短纤维之间相互搭接形成的交错结构。由于多根纳米短纤维之间相互搭接形成的交错结构且微纤维的尺寸较小,使得微纤维态止血材料处于更加蓬松的状态,从而可以进一步增大微纤维态止血材料的比表面积。
本发明中的微纤维,其意指尺寸较小的纤维。具体地,微纤维的直径为1μm~500μm,可以在1μm~400μm之间,可以在1μm~300μm之间,可以在1μm~200μm之间,可以在1μm~100μm之间;长度一般为0.5mm~10mm,可以在0.5mm~8mm之间,可以在0.5mm~5mm之间,可以在1mm~5mm之间。由于本发明的微纤维的长度在0.5mm~10mm之间,从而容易塑形为适合于各种创面以进行更有效的止血,比如簇状、团状等。如果微纤维的尺寸小于0.1mm,例如粉状的止血材料,在出血较多的情况下容易被冲走。
另外,本发明的微纤维的长度与直径的比值可以在1~10000的范围内,优选在5~8000的范围内,还可以在8~5000的范围内;当在5~8000的范围内时,可以使微纤维态止血材料同时保持较高的蓬松度和比表面积,增强与创面之间的粘附强度以及止血效果,并且方便医生通过镊子直接夹持止血材料施用到创面上。
<微纤维态止血材料>
如图1所示,本发明的微纤维态止血材料包括微纤维。微纤维态止血材料的堆密度小于0.03g/cm3,优选为0.01~0.025g/cm3。微纤维态止血材料的堆密度小,具有高度蓬松的特性以及超高的比表面积。所述微纤维态止血材料的比表面积可以为4m2/g~50m2/g,可以为6m2/g~30m2/g。并且微纤维态止血材料具有大于1500%,优选在2000%~2500%之间的吸水率,具有高吸水性能。
由于本发明的微纤维态止血材料具有超高的比表面积和超高的吸水性能,从而能够在出血创面迅速吸收血液中的水分,可以提高血液中红细胞、凝血因子等的浓度,加速内源性凝血机制,提高止血效果。
进一步地,本发明的微纤维态止血材料还包含有药物。优选地,所述药物包括凝血因子、生长因子中的一种或两种的组合。通过加载药物,不仅可以提高材料的止血性能,还同时具备促进创口快速愈合、防粘连等性能。
本发明的微纤维态止血材料,相对于粉状的止血材料而言,微纤维态的结构更容易形成有一定强度的物理压迫,避免粉状的材料在出血较多的情况下容易被冲走的情况。
第二实施方式
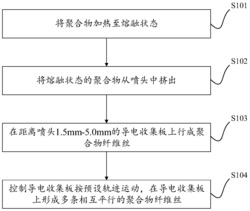
本发明的第二实施方式提供了一种微纤维态止血材料的制备方法,包括以下步骤:
静电纺丝步骤:通过静电纺丝制备所述纳米纤维材料;
交联步骤:在交联剂的存在下对所述纳米纤维材料进行交联处理,得到交联的纳米纤维材料;
剪切步骤:对所述交联的纳米纤维材料进行剪切处理。
本文中所述的“交联”与“交联改性”具有相同或相似的含义,在“交联”的过程中,可以附带有“改性”的一些特点,本发明中为了简便,可以使用“交联”代替“交联改性”。
<静电纺丝步骤>
所述静电纺丝步骤中,预先准备纤维原料,将纤维原料溶于合适的溶剂中,制备成一定浓度的纤维原料的纺丝原液。其中,所述纤维原料可以为第一实施方式中的聚合物材料。对于形成溶液的溶剂种类的具体浓度没有特别的限定,只要能够满足后续静电纺丝工艺的要求即可。举例而言,合适的溶剂可以为三氟乙醇、六氟异丙醇、三氟乙酸、环己酮、丙酮、丁酮、四氢呋喃、氯仿、冰乙酸、甲酸、丙酸或水中的一种或两种以上的组合。
静电纺丝过程中可以通过调节纺丝参数制备所需的纳米纤维材料。例如电压、挤出流量和电场接收距离、纺丝环境等。优选地,本发明中所述静电纺丝工艺参数可以为:压力为10~40kV,溶液挤出流量为0.1~15mL/h,电场接收距离为5~30cm,纺丝环境相对温度在60%以下,环境温度为10~40℃。
另外,可以考虑在纺丝原液或者在电纺过程中加载药物,所述药物可以包括凝血因子、生长因子等中的一种或两种的组合。不仅可以提高材料的止血性能,还同时具备促进伤口快速愈合、防粘连等性能。
<交联步骤>
所述交联步骤中,所选用的交联剂包括碳化二亚胺、N-羟基琥珀酰亚胺、京尼平、醛类化合物中的一种或两种以上的组合。
考虑到交联剂对生物体毒性大小和交联效果,交联剂可以选用碳化二亚胺和/或N-羟基琥珀酰亚胺。按照本发明的研究发现,通过使用N-羟基琥珀酰亚胺,可以进一步提高碳化二亚胺的交联效果。因此,化学交联剂优选为碳化二亚胺和N-羟基琥珀酰亚胺的组合。更优选地所述碳化二亚胺与N-羟基琥珀酰亚胺的质量比为1~4:1。
一般而言,交联改性是在溶液中进行的,本发明中对进行交联改性处理的溶剂不做具体限定,只要能够满足交联改性反应的需求即可。在本发明中,进行交联改性处理的溶剂可以是不同质量比的醇与水的混合溶液。其中,醇优选为乙醇,更优选地,乙醇与水的质量比在70%以上。
进一步地,可以通过调整交联处理的反应条件、交联剂的用量从而控制交联改性的情况。例如交联处理温度、交联处理时间、交联剂与纳米纤维材料的质量比、交联剂的质量与溶剂的体积之比等。另外,可以通过调控交联程度,从而满足不同创伤或临床手术对降解周期的要求。
本发明中所述交联步骤中的反应条件可以是,交联处理温度在1~50℃之间,优选在4~30℃之间。交联处理时间在1~72h之间,优选在6~24h之间。所述化学交联剂的质量与纳米纤维材料的质量比为0.1~3:1,优选为0.5~2:1。化学交联剂的质量与溶剂的体积之比为0.1~10:100,其中优选1~5:100。可以通过控制上述反应条件进一步控制交联程度。
另外,本发明中,也可以先对纤维原料进行交联处理,然后再采用静电纺丝技术进行处理,优选为先采用静电纺丝技术进行处理,再对纳米纤维材料进行交联处理。
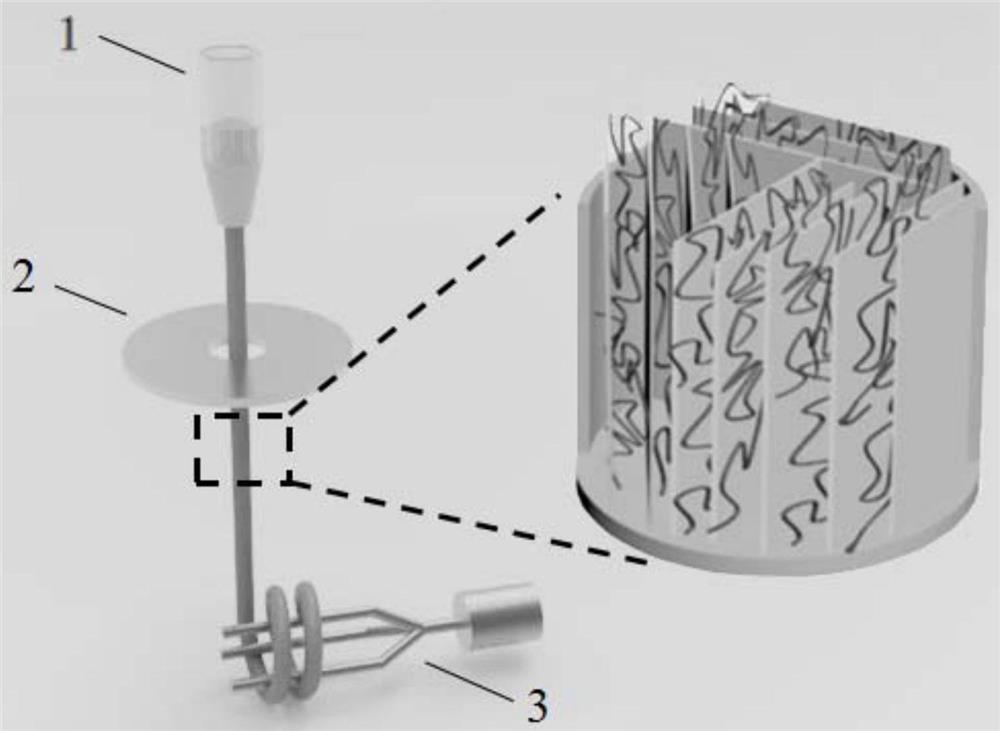
<剪切步骤>
本发明的剪切步骤中,所述剪切处理包括预剪切步骤,具体地,所述预剪切步骤为在5000~10000rpm/min的转速下对所述交联的纳米纤维材料进行初步剪切处理,得到预剪切物。一般而言,预剪切步骤的处理时间为5~30mim。通过预剪切处理,能够将交联体系初步切碎得到均匀的小片状材料。
进一步地,所述剪切处理还包括高速剪切步骤,所述高速剪切步骤中,为了最终得到的微纤维形成较均一的微纤维态,高速剪切步骤的转速和时间较为关键;具体地,高速剪切步骤的转速可以在20000~40000rpm/min,优选在25000~30000rpm/min之间;高速剪切步骤的时间为1~10min,更优选为3~5min。
如果高速剪切步骤的转速小于20000rpm/min,高速剪切步骤时间小于1min,一定程度上会使止血材料大多数呈现小片状,以及剪切不均匀的现象;如果转速大于40000rpm/min,高速剪切步骤的时间大于10min,一定程度上会使止血材料呈现为粉末状,而非微纤维态,而粉末状的止血材料其蓬松度低,形态密实,并且在临床使用操作和止血效果上不能达到微纤维态止血材料的同等水平。
进一步地,所述剪切处理是在通入流动气体的状态下进行的。具体而言,所述剪切处理在容器中进行,且所述容器中通入有流动气体。所述容器可以是密闭的,或者非密闭的。举例而言,本发明的剪切处理可以是利用特定的剪切机进行处理的。剪切机可以具有用于盛放所述交联的纳米纤维材料的容器。可以在容器中通入流动气体,进而对所述交联的纳米纤维材料进行剪切。经剪切后得到的纳米短纤维中,至少部分纳米短纤维相互搭接形成微纤维,且微纤维能够蓬松的分布在所述容器的内部空间中。
优选地,本发明剪切机可以通过充气的方式向容器中通入流动气体。可以是连续充气也可以是间歇充气,还可以是循环充气的方式。本发明中的流动气体可以是形成对流的气体,或者是形成扰动的气体等。
本发明的容器的材料优选为非金属材质,例如:有机玻璃、四氟乙烯等非金属材质。这是由于高速剪切过程中,容器与电机相连,金属材质容易导热,高温容易使微纤维发生溶解变性,从而在一定程度上影响微纤维的性能,进而影响微纤维态止血材料的性能。因此,选用非金属材质,可以避免影响微纤维态止血材料的性能。另外,为了便于观察剪切效果,优选为有机玻璃材质。
由于在剪切过程中通入流动气体,因此剪切的更加均匀,并且能够进一步使得微纤维获得更高的蓬松度,从而进一步增大微纤维的比表面积。
<洗脱步骤、冷冻干燥步骤>
在所述交联步骤与剪切步骤之间还可以包括:洗脱步骤和/或冷冻干燥步骤。
进行洗脱的目的是去除未反应的交联剂。具体地,所述洗脱步骤可以包括:在0~20℃的低温下利用洗脱剂,通过浓度梯度法洗脱所述交联的纳米纤维材料,以去除所述未反应的交联剂。所述洗脱剂包括醇类与水的混合液,优选包括乙醇和水的混合液,更优选地,乙醇和水的混合液中乙醇的质量分数在70%以上。另外,本发明中的洗脱剂与交联处理中所采用的溶剂可以相同,也可以不同。
进一步地,本发明中可以利用不同浓度的醇-水溶液,采用浓度梯度法进行多次洗脱。优选地,洗脱条件为:洗脱温度为1~20℃,优选4~10℃;洗脱时间为0.1~5h,优选0.5~2h,重复3~5次。
进行冷冻干燥的目的是除去交联处理中的多余的溶剂和洗脱剂,同时有助于提高纳米纤维材料的孔隙率。所述冷冻干燥的具体步骤为,将洗脱后的纳米纤维材料放入容器中,在-20~-80℃下预冷1~3h,然后将容器转移至冷冻干燥机中进行冻干,冻干的温度为-10~40℃,优选-10~30℃;在1~100pa,优选20~40pa真空度下冻干6~72h,优选12~24h。
通过采用本发明的制备方法制备得到的微纤维态止血材料,相对于市售的胶原类微纤维止血粉,生产加工工艺更为简单,成本更低,原材料的选择来源更为广泛,更加符合工业化生产的要求。
另外,可以将剪切后得到的微纤维密封包装,进行Co-60γ射线辐照灭菌处理。具体地,所述密封包装要求在干燥环境下,环境湿度在30%以下快速封装;Co-60γ射线辐照剂量为15~30kGY。
本发明制备的微纤维态止血材料,使用时无需提前准备,只需将其从包装中取出,即可用于创面,节省了宝贵的抢救时间,方便和简化了操作,同时产品携带和保存更为简便。
第三实施方式
本发明的第三实施方式还提供一种止血制品,包括根据本发明第一实施方式的微纤维态止血材料,或者根据本发明第二实施方式的微纤维态止血材料的制备方法制备得到的微纤维态止血材料。
本发明的止血制品可用于在组织渗血、毛细血管出血、小动脉出血、腔隙渗血时的止血和修复,和/或,用于烧伤、创伤、外科手术创口的止血和修复,具有广阔的应用前景。当应用在腔隙渗血时的止血和修复时,可以借助于辅助配套器械将其应用于腔隙等部位的渗血,或者医生可以根据经验,将本产品与其他市售产品联用,比如止血海绵,止血纱布等产品,从而达到更好的止血效果。
本发明的止血制品,由于微纤维态止血材料具有良好的粘附性能,在伤口表面形成粘附能力较好的凝胶,进行良好的物理封堵而实现压迫止血。同时,由于具有超高比表面积以及亲水性的聚合物材料的选择,可以使得材料具有高亲水性能。在出血创面能迅速吸收血液中的水分,从而提供血液中红细胞、凝血因子等的浓度,加速内源性凝血机制,提高止血效果。
实施例
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例1
(1)将羧甲基壳聚糖(CMCH)溶于纯化水中,其中,羧甲基壳聚糖的质量浓度为5%(g/mL),搅拌溶解得到均匀的聚合物溶液,即为纺丝原液。将聚合物溶液置于静电纺丝注射器中,调节微量注射泵的速率为2mL/h,调节高压发生器的电压为30kV,调节接收装置的接收距离为15cm,纺丝环境相对湿度设为30%,环境温度为40℃,进行静电纺丝。通过高压静电纺丝技术,制备得到由纤维丝交织而成且具有多孔结构的纳米纤维材料。
(2)在500mL反应器中,加入150mL无水乙醇溶液,再加入50mL水溶液和4mL戊二醛溶液后搅拌混匀,将2g纳米纤维材料放入反应器中,40℃下进行交联改性,处理24h,得到交联的纳米纤维材料。
(3)配置乙醇质量分数为70%的乙醇-水溶液,在4℃的低温下预冷一段时间3h后,将交联的纳米纤维材料转移至上述乙醇质量分数为70%的乙醇-水溶液中进行洗脱;洗脱1h后,再次转移至乙醇质量分数为95%的4℃的乙醇-水溶液中;洗脱1h后,再转移至乙醇质量分数为70%的4℃的乙醇-水溶液中,洗脱1h。利用浓度梯度法进行洗脱,以去除未反应的戊二醛,重复3次。
(4)将洗脱后的纳米纤维材料放入洁净的容器在-80℃下预冷3h,然后将容器转移至冷冻干燥机中进行冻干,设置冻干的温度为-10℃下干燥3h,然后再在20℃下干燥24h,真空度设置为20pa。
(5)将冷冻干燥后的纳米纤维材料,放入高速剪切机中,设置转速为5000rpm/min,处理时间为10min,进行初步剪切,得到片状的预剪切物;然后将转速设置为25000rpm/min,处理时间为10min,得到微纤维。
(6)将剪切后得到的微纤维密封包装,进行25kGY的Co-60γ射线辐照灭菌处理后得到微纤维态的止血产品。
实施例2
(1)将胶原(Collagen)溶于三氟乙醇中,其中,胶原的质量浓度为7%(g/mL),搅拌溶解得到均匀的聚合物溶液,即为纺丝原液。将所述聚合物溶液置于静电纺丝注射器中,调节微量注射泵的速率为5mL/h,调节高压发生器的电压为25kV,调节接收装置的接收距离为12cm,纺丝环境相对湿度设为40%,环境温度为30℃,进行静电纺丝,通过高压静电纺丝技术,制备得到由纤维丝交织而成且具有多孔结构的纳米纤维材料。
(2)在500mL反应器中,加入225mL无水乙醇溶液,再加入25mL水溶液,搅拌混匀,然后称取3.57g碳化二亚胺和1.43gN-羟基琥珀酰亚胺在常温下溶解于上述乙醇-水溶液中,将5g纳米纤维材料放入反应器中,25℃下进行交联改性,处理12h,得到交联的纳米纤维材料。
(3)配置乙醇质量分数为70%的乙醇-水溶液,在4℃的低温下预冷一段时间3h后,将交联的纳米纤维材料转移至上述乙醇质量分数为70%的乙醇-水溶液中进行洗脱;洗脱1h后,再次转移至乙醇质量分数为95%的4℃的乙醇-水溶液中;洗脱1h后,再转移至乙醇质量分数为70%的4℃的乙醇-水溶液中洗脱,洗脱1h。进行利用浓度梯度法进行洗脱,以去除未反应的碳化二亚胺和N-羟基琥珀酰亚胺,重复3次。
(4)将洗脱后的纳米纤维材料放入洁净的容器中在-80℃下预冷3h,然后将容器转移至冷冻干燥机中进行冻干,设置冻干的温度为10℃下干燥3h,然后再在20℃下干燥24h,真空度设置为30pa。
(5)将冷冻干燥后的纳米纤维材料,放入高速剪切机中,设置转速为8000rpm/min,处理时间为5min,进行初步剪切,得到片状的预剪切物;然后将转速设置为30000rpm/min,处理时间为5min,得到微纤维。
(6)将剪切后得到的微纤维密封包装,进行25kGY的Co-60γ射线辐照灭菌处理后得到微纤维态的止血产品,电镜图如图1和图2所示。
实施例3
(1)将羟丙基甲基纤维素(HPMC)材料溶于六氟异丙醇与水的混合溶液中,其中,羟丙基甲基纤维素质量浓度为10%(g/mL),搅拌溶解得到均匀的聚合物溶液,即为纺丝原液。同时按照羟丙基甲基纤维素20质量%的纤维蛋白原(凝血因子)加入上述聚合物溶液中进行溶解。将溶解有纤维蛋白原的聚合物溶液置于静电纺丝注射器中,调节微量注射泵的速率为5mL/h,调节高压发生器的电压为38kV,调节接收装置的接收距离为10cm,纺丝环境相对湿度设为30%,环境温度为40℃,进行静电纺丝。通过高压静电纺丝技术,制备出复合凝血因子的由纤维丝交织而成且具有多孔结构的纳米纤维材料。
(2)在500mL反应器中,加入160mL无水乙醇溶液,再加入40mL水溶液和8mL戊二醛溶液后,并将PH值调节至酸性(pH<4),搅拌混匀,将6g纳米纤维材料放入反应器中,35℃下进行交联改性,处理48h,得到交联的纳米纤维材料。
(3)配置乙醇质量分数为70%的乙醇-水溶液,在4℃的低温下预冷一段时间3h后,将交联的纳米纤维材料转移至上述乙醇质量分数为70%的乙醇-水溶液中进行洗脱;洗脱1h后,再次转移至乙醇质量分数为95%的4℃的乙醇-水溶液中;洗脱1h后,再转移至乙醇质量分数为70%的4℃的乙醇-水溶液中,洗脱1h。利用浓度梯度法进行洗脱,以去除未反应的戊二醛,重复3次。
(4)将洗脱后的纳米纤维材料放入洁净的容器在-80℃下预冷3h,然后将容器转移至冷冻干燥机中进行冻干,设置冻干的温度为30℃下干燥3h,然后再在20℃下干燥24h,真空度设置为40pa。
(5)将冷冻干燥后的纳米纤维材料,放入高速剪切机中,设置转速为10000rpm/min,处理时间为5min,进行初步剪切,得到片状的预剪切物;然后将转速设置为40000rpm/min,处理时间为1min,得到微纤维。
(6)将剪切后得到的微纤维密封包装,进行25kGY的Co-60γ射线辐照灭菌处理后得到微纤维态的止血产品。
性能测试
比表面积测试
测试方法:取待测产品放入分析仪器的样品管中,其中,分析仪器为快速全自动比表面积和孔径分析仪,型号为美国康塔NOVA 4200e。在低温(液氮浴)条件下,向样品管内通入一定量的吸附质气体(N2),根据吸附前后气体体积的变化来确定被测样品对吸附质分子(N2)的吸附量;参考国家标准GB/T24533-2009—气体吸附BET原理来测定固态物质的比表面积。
比表面积的计算方式为:放到气体环境中的样品中,其物质表面(颗粒外部和内部空隙的表面积)在低温下将发生物理吸附。当吸附达到平衡时,测量平衡吸附压力的吸附气体量,根据BET方程式计算出试样单分子层吸附量,从而计算出试样的比表面积。其中,BET方程式为:
式中:
P——吸附质分压,单位为Pa;
P0——吸附剂饱和蒸汽压,单位为Pa;
V——样品实际吸附量,单位为cm3;
Vm——单层饱和吸附量,单位为cm3;
C——与样品吸附能力相关的常数。
以实施例1-3的微纤维态止血纤维产品进行测试,测试结果如表1所示。
表1
由表1可以看出,本发明的微纤维态止血材料具有很高的比表面积。
饱和吸水率测试
测试方法:将一定质量的样品(m1)置于培养皿内,加入预热至(37±1)℃的0.9%生理盐水,生理盐水的质量为供试材料的40倍。将培养皿移入干燥箱内,在(37±1)℃下保持30min。用镊子取出样品,悬垂30s,用电子天平准确称量m2,平行测定3次。通过计算得到饱和吸水率(X),饱和吸水率(X)的计算公式为:X=(m2-m1)/m1×100%。
以实施例1-3的微纤维态止血纤维产品与三种不同的市售产品1-3(均为市场上销量较好的止血材料)进行测试,测试结果如表2所示,其中,n为平行测定次数。
表2饱和吸水率测试结果(n=3)
从上表2的数据表明,按照本发明的产品吸水性能良好,所有实施例的产品的饱和吸水率能达1500%以上,明显优于其他市售产品。并且,按照实施例2制备的产品的饱和吸水率最高,可达材料自身体重的24.7倍。
堆密度测试
测试方法:将材料自由落入体积为V(cm3),质量为m1(g)的量筒内,使材料自由堆叠达到既定体积,然后称取量筒与材料的总体质量m2(g),则堆密度(g/cm3)=(m2-m1)/V。
以实施例1-3的产品与市售产品1进行测试,结果如表3所示,其中,n为平行测试次数。
表3堆密度测试(n=3)
由表3可以看出,本发明的产品具有良好的蓬松度,从而可以在伤口表面形成一定的支架结构。当微纤维态止血材料快速吸收血液中的水分后,立即膨胀形成凝胶状,对创面进行物理封堵止血。同时血浆中的有效止血成分浓度提升后,可以更好的促进凝血机制的发生,起到双重止血功能。
止血有效性测试
测试方法:采用兔子肝脏渗血模型,剪去腹部兔毛,标准的正中开腹,游离、暴露肝脏;在肝脏相同部位形成10×10×2mm的伤口;用纱布清理创面,用相同重量的止血材料覆盖创口表面,并在上面覆盖明胶海绵,按压30s,移除海绵并观察创口渗血情况。记录止血时间,评价止血有效性。
以本发明实施例2制得的产品(实验组)进行测试,同时采用对照产品(市售产品1)作为阳性对照,实验组和对照组平行组数为n=8。
结果如图3所示,对照产品(市售产品1)的止血时间为211.2s;本发明实施例2制得的微纤维态止血材料的止血时间为161.5s;P值=0.023,P值<0.05,有显著性差异。因此,本发明的产品的止血时间显著小于市售产品1的止血时间。
安全性及降解性测试
测试方法:以本发明实施例2的产品作为试验材料(实验组),以市售产品1作为对照材料(对照组)。
采用大鼠肝脏原位止血植入模型。在大鼠腹部备毛,标准的正中开腹,游离、暴露肝脏;选择其中最大的一片肝叶,剪出一个10×10×2mm的创面。称0.15g止血材料放至伤口表面,进行原位止血并植入,缝合关闭,术后抗生素护理三天。到预先设定的观察周期(术后7天、术后14天、术后28天)后,解剖拍照记录,并取组织送病理分析,病理分析评价植入位点材料的生物组织相容性。
病理结果显示,如图4所示,本发明的微纤维态止血材料在28天时已降解完全。而市售产品1在28天时还能看到部分材料未完全降解吸收。并且,实验组的组织验证刺激也低于对照组。因此,本发明的微纤维态止血材料具有良好的组织生物相容性。另外,在实验的过程中,能够观察到本申请的微纤维态止血材料具有更加优异的粘附性能。
本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
微纤维态止血材料及其制备方法和止血制品专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











动态评分
0.0