

IPC分类号 : C08J3/24I,C08J3/09I,C08L47/00I,C08L71/02I,C08L75/08I,C08K5/06I,C08K5/02I,C06B23/00I,C06B45/10I,C06D5/00I







专利摘要
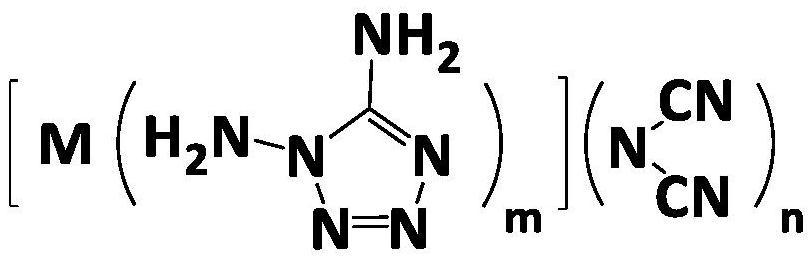
本发明涉及一种可溶解的三维交联弹性体及其制备方法和处理方法,属于复合固体推进剂技术领域。本发明采用端三唑基预聚物与卤代物反应生成三唑鎓基团三维交联的聚合物弹性材料。该弹性体具有稳定的化学交联网络。与目前常规共价键交联聚合物弹性体不同,三唑鎓离子交联的聚合物弹性体遇一元卤代化合物可以发生离子交换反应,使得三维交联网络解离。利用该类弹性体制备复合固体推进剂可实现聚合物交联网络解离,弹性体由固态变为液态,实现废旧复合固体推进剂的高效回收、利用。
权利要求
1.一种可溶解的三维交联弹性体,其特征在于:该三维交联弹性体由端三唑基预聚物粘合剂和固化剂反应制备;
所述端三唑基预聚物粘合剂是指在一个高分子链结构中具有2个或2个以上端三唑基基团的高分子预聚物,为端三唑基聚丁二烯、端三唑基聚乙二醇、端三唑基聚环氧乙烷四氢呋喃共聚醚、端三唑基聚叠氮缩水甘油醚、端三唑基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚中的一种或其混合物;
所述固化剂为分子结构中含有2个或2个以上端溴原子或端碘原子的化合物,选自1,2-二溴丙烷、1,3-二溴丙烷、1,4-二溴丁烷、1.5-二溴戊烷、1,5-二碘丁烷、1,6-二碘代己烷、1,6-二溴代己烷、1,7-二溴代庚烷、1,7-二碘代庚烷、1,8-二碘代辛烷、1,8-二溴代辛烷、1,6-二碘代十二氟己烷、二溴代异丁酸乙二醇酯、1,2,3-三溴丙烷、三溴甲基丙烷、三碘甲基丙烷、三溴新戊醇、4,4’-二碘-1,1’-联苯、1,4-二溴代萘中的一种或两种以上的混合物;
所述端三唑基预聚物粘合剂中三唑官能团的摩尔数与所述固化剂中卤素的摩尔数的比为0.8-1.2:1。
2.根据权利要求1所述的一种可溶解的三维交联弹性体,其特征在于:所述端三唑基预聚物粘合剂分子量范围为3000~10000g/mol。
3.一种根据权利要求1所述的可溶解的三维交联弹性体的制备方法,其特征在于,步骤包括:
(1)将端三唑基预聚物粘合剂加入到反应器中;
(2)将固化剂加入到步骤(1)的反应器中,搅拌均匀;
(3)将步骤(2)所得混合物真空浇铸至模具中;
(4)将模具放入烘箱中,加热温度至80-100℃,模具混合物中三唑基团与卤代化合物反应,生成三唑鎓交联的聚合物弹性体,即得到三维交联弹性体。
4.一种根据权利要求1所述的可溶解的三维交联弹性体的处理方法,其特征在于,步骤如下:
将三维交联弹性体浸泡于溶剂中,三维交联弹性体逐渐溶解变成溶液,溶解时间为5-6天,溶剂具体为1-溴代戊烷、1-溴代己烷、1-溴代辛烷、1-碘代戊烷、1-碘代己烷或1-碘代辛烷。
说明书
技术领域
本发明涉及一种可溶解的三维交联弹性体及其制备方法和处理方法,属于复合固体推进剂技术领域。
背景技术
复合固体推进剂作为固体火箭发动机的动力来源,其不仅要满足固体火箭发动机能源需求;同时作为工程构件,复合固体推进剂还需在较宽温度范围内具有一定的力学性能,满足不同工作环境要求。复合固体推进剂是以聚合物交联网络弹性体为连续相、以固体填料为分散相的复合材料,其中聚合物连续相是承载复合固体推进剂力学性能的基体;力学性能稳定、良好的聚合物弹性体是制备高性能复合固体推进剂的前提保障。
为确保复合固体推进剂力学性能稳定性,利用具有稳定三维交联网路结构的聚合物弹性体是制备复合固体推进剂的首选材料。因此,自二十世纪四十年代以来,复合固体推进剂先后经历的聚硫橡胶(PSR)复合推进剂、聚丁二烯丙烯酸(PBAA)复合推进剂、聚丁二烯丙烯酸丙烯腈(PBAN)复合推进剂、端羧基聚丁二烯(CTPB)复合推进剂、端羟基聚丁二烯(HTPB)复合推进剂、硝酸酯增塑的聚醚复合推进剂(NEPE)以及含能增塑剂增塑的叠氮聚醚复合推进剂等均是采用共价键三维交联的聚合物网络作为复合固体推进剂连续相基体材料。
由于共价键三维交联聚合物网络结构稳定、难以解离,对废旧复合固体推进剂的处理变得异常困难。目前,通常采用的露天焚烧法、高压水切割法、液氮切割法及其它萃取法等手段处理废旧复合固体推进剂引发严重的环境污染、安全隐患及高成本等问题。对复合固体推进剂组分中的粘合剂体系进行改性,使其三维化学交联聚合物网络结构易于分解或解离,实现废旧复合固体推进剂组分高效回收利用是降低复合固体推进剂制造成本、消除废旧复合固体推进剂后处理问题的重要途径。
发明内容
本发明目的是为了解决常规共价键三维交联聚合物弹性体难以解离的问题,提出了一种溶剂可溶、离子键交联的聚合物弹性体及其制备方法和处理方法。
本发明以端三唑基预聚物为粘合剂,以小分子多元卤代化合物为固化剂,通过预聚物中端三唑基团与固化剂发生烷基化反应,生成三唑鎓离子交联点,形成离子键三维交联的聚合物网络结构。遇一元卤代化合物时,该三唑鎓离子三维交联结构发生解离,固态聚合物弹性体变为溶胶液体。
本发明的目的是通过以下技术方案实现的。
本发明的一种可溶解的三维交联弹性体,该三维交联弹性体包括端三唑基预聚物粘合剂和固化剂,粘合剂中三唑官能团的摩尔数与固化剂中卤素的摩尔数的比为0.8-1.2:1;
其中,固化剂为多官能度端卤素化合物;
端三唑基预聚物粘合剂为端三唑基聚丁二烯(Triazole-terminatedpolybutadiene,TTPB)、端三唑基聚醚(Triazole-terminated polyether,TTPE)中的一种或其混合物;
端三唑基预聚物具体指端三唑基聚丁二烯、端三唑基聚乙二醇、端三唑基聚环氧乙烷四氢呋喃共聚醚、端三唑基聚叠氮缩水甘油醚、端三唑基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚;
端三唑基聚丁二烯的制备方法为:
(1)将端炔基聚丁二烯溶于正庚烷溶剂中,置于带有机械搅拌和氮气保护的容器中,加入抗坏血酸钠和CuSO4,搅拌均匀;
(2)将叠氮化物加入到步骤(1)的容器中,得到混合物;叠氮化物为苄基叠氮或2-叠氮基乙酸甲酯;
(3)将步骤(2)的混合物在30~80℃、搅拌条件下反应3~6小时,冷却,去除氮气保护;
(4)将步骤(3)得到的反应混合物用蒸馏水萃取三次,旋蒸、干燥后得到端三唑基聚丁二烯预聚物。
所述步骤(1)中,所用端炔基聚丁二烯、正庚烷溶剂、抗坏血酸钠、CuSO4的质量比为100:180-260:1-2:0.4-0.6;
所述步骤(2)中,加入叠氮化物的摩尔数与步骤(1)中端炔基聚丁二烯的端炔基基团的摩尔数比为1:1。
端三唑基聚乙二醇的制备方法为:
(1)将端叠氮基聚乙二醇溶于四氢呋喃溶剂中,置于带有机械搅拌和氮气保护的容器中,加入抗坏血酸钠和CuSO4,搅拌均匀;
(2)将丙炔醇加入步骤(1)的容器中;
(3)将步骤(2)混合溶液在30~60℃、搅拌条件下反应3~6小时,冷却,去除氮气保护;
(4)将步骤(3)得到的反应混合物用蒸馏水萃取三次,旋蒸、干燥后得到端三唑基聚乙二醇。
所述步骤(1)中,所用端叠氮基聚乙二醇、四氢呋喃、抗坏血酸钠、CuSO4的质量比为100:180-600:0.5-1.5:0.3-0.5;
所述步骤(2)中,加入丙炔醇的摩尔数与步骤(1)中端叠氮基聚乙二醇的端叠氮基团的摩尔数比为1:1。
端三唑基聚环氧乙烷四氢呋喃共聚醚的制备方法为:
(1)将端叠氮基聚环氧乙烷四氢呋喃共聚醚溶于四氢呋喃溶剂中,置于带有机械搅拌和氮气保护的容器中,加入抗坏血酸钠和CuSO4,搅拌均匀;
(2)将丙炔醇加入步骤(1)的容器中;
(3)将步骤(2)混合溶液在30~60℃、搅拌条件下反应3~6小时,冷却,去除氮气保护;
(4)将步骤(3)得到的反应混合物用蒸馏水萃取三次,旋蒸、干燥后得到端三唑基聚环氧乙烷四氢呋喃共聚醚。
所述步骤(1)中,所用端叠氮基聚环氧乙烷四氢呋喃共聚醚、四氢呋喃、抗坏血酸钠、CuSO4的质量比为100:180-600:0.5-1.5:0.3-0.5;
所述步骤(2)中,加入丙炔醇的摩尔数与步骤(1)中端叠氮基聚环氧乙烷四氢呋喃共聚醚的端叠氮基团的摩尔数比为1:1。
端三唑基聚叠氮缩水甘油醚的制备方法为:
(1)将端羟基聚叠氮缩水甘油醚、无水四氢呋喃溶剂放入带有机械搅拌和冷凝管装置的容器中;
(2)室温下,将过量的甲苯二异氰酸酯(TDI)加入步骤(1)的容器中,搅拌均匀后升温至50~65℃反应6~8小时,降至室温;
(3)往步骤(2)容器中加入与步骤(2)所用TDI的异氰酸酯基等摩尔量的1-苯甲基-4-羟甲基三唑,搅拌均匀后升温至50~65℃继续反应6~8小时;
(4)旋蒸去除步骤(3)中四氢呋喃溶剂,得到端三唑基聚叠氮缩水甘油醚粗品;
(5)将步骤(4)所得端三唑基聚叠氮缩水甘油醚粗品溶于DMF溶剂中,然后加入蒸馏水振荡、静置分层,取油相减压蒸馏后得到端三唑基聚叠氮缩水甘油醚。
所述步骤(2)中,过量TDI的含义为:当步骤(1)中端羟基聚叠氮缩水甘油醚的羟基摩尔数为1时,步骤(2)中TDI的摩尔数不小于1.5。
端三唑基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚的制备方法为:
(1)将端羟基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚、无水四氢呋喃溶剂放入带有机械搅拌和冷凝管装置的容器中;
(2)室温下,将过量的TDI加入步骤(1)的容器中,搅拌均匀后升温至50~65℃反应6~8小时,降至室温;
(3)往步骤(2)容器中加入与步骤(2)所用TDI的异氰酸酯基等摩尔量的1-苯甲基-4-羟甲基三唑,搅拌均匀后升温至50~65℃继续反应6~8小时;
(4)旋蒸去除步骤(3)中四氢呋喃溶剂,得到端三唑基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚粗品;
(5)将步骤(4)所得端三唑基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚粗品溶于DMF溶剂中,然后加入蒸馏水振荡、静置分层,取油相减压蒸馏后得到端三唑基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚。
所述的步骤(2)中,过量的TDI的含义为:当步骤(1)中端羟基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚的羟基摩尔数为1时,步骤(2)中TDI的摩尔数不小于1.5。
端三唑基预聚物是指在一个高分子链结构中具有2个或2个以上端三唑基基团的高分子预聚物,预聚物分子量范围为3000~10000g/mol;
多官能度端卤素化合物是指在一个分子结构中含有2个或2个以上卤原子的化合物,卤原子为溴原子或碘原子;
多官能度端卤素化合物具体为1,2-二溴丙烷、1,3-二溴丙烷、1,4-二溴丁烷、1.5-二溴戊烷、1,5-二碘丁烷、1,6-二碘代己烷、1,6-二溴代己烷、1,7-二溴代庚烷、1,7-二碘代庚烷、1,8-二碘代辛烷、1,8-二溴代辛烷、1,6-二碘代十二氟己烷、二溴代异丁酸乙二醇酯、1,2,3-三溴丙烷、三溴甲基丙烷、三碘甲基丙烷、三溴新戊醇、4,4’-二碘-1,1’-联苯、1,4-二溴代萘中的一种或两种以上的混合物。
一种可溶解的三维交联弹性体的制备方法,其具体实施步骤如下:
(1)将端三唑基预聚物粘合剂加入到反应器中;
(2)将固化剂加入到步骤(1)的反应器中,搅拌均匀;
(3)将步骤(2)所得混合物真空浇铸至模具中;
(4)将模具放入烘箱中,加热温度至80-100℃,模具混合物中三唑基团与卤代化合物反应,生成三唑鎓交联的聚合物弹性体,即得到三维交联弹性体。
一种可溶解的三维交联弹性体的处理方法,其具体实施步骤如下:
将得到的三维交联弹性体浸泡于溶剂中,三维交联弹性体逐渐溶解变成溶液,溶解时间为5-6天;溶剂为一元卤代物,具体为1-溴代戊烷、1-溴代己烷、1-溴代辛烷、1-碘代戊烷、1-碘代己烷或1-碘代辛烷。
有益效果
本发明采用端三唑基预聚物粘合剂与卤代物反应生成三唑鎓基团三维交联的聚合物弹性体材料。该弹性体具有稳定的化学交联网络。与目前常规共价键交联聚合物弹性体不同,三唑鎓离子交联的聚合物弹性体遇一元卤代化合物可以发生离子交换反应,使得三维交联网络解离。利用该类弹性体制备复合固体推进剂可实现其聚合物交联网络解离,弹性体由固态变为液态,实现废旧复合固体推进剂的高效回收、利用。
具体实施方式
下面通过实施例对本发明作进一步说明,但实施例并不限制本发明的保护范围。
列举实施方式中所用端炔基聚丁二烯的数均分子量为3000g mol
端三唑基聚丁二烯的制备方法为:
1)将100g端炔基聚丁二烯溶于200mL正庚烷溶剂中,置于配有搅拌装置、氮气保护的500mL三口烧瓶中,加入1.31g抗坏血酸钠、0.527g CuSO4,搅拌均匀;
2)将与步骤1)中端炔基基团等摩尔当量的8.788g苄基叠氮加入步骤1)的容器中;【或2-叠氮基乙酸甲酯7.597g】
3)将步骤2)的混合溶液在50℃、搅拌条件下反应4小时,冷却,除去氮气保护;
4)将步骤3)所得反应混合物用50mL蒸馏水萃取三次,旋蒸、干燥后得到约95g端三唑基聚丁二烯预聚物。
产物红外分析表明,3306cm
列举实施方式中所用端叠氮基聚乙二醇的数均分子量4000g mol
端三唑基聚乙二醇的制备方法为:
1)将40g端叠氮基聚乙二醇溶于200mL四氢呋喃溶剂中,置于配有机械搅拌、氮气保护的500mL三口烧瓶中,加入0.37g抗坏血酸钠、0.15g CuSO4,搅拌均匀;
2)将1.04g丙炔醇加入步骤1)的容器中;
3)将步骤2)的混合溶液在40℃、搅拌条件下反应4小时,冷却,除去氮气保护;
4)将步骤3)的反应混合物用蒸馏水萃取三次,旋蒸、干燥后得到38g端三唑基聚乙二醇。
产物红外分析表明,2100cm
列举实施方式中所用端叠氮基聚环氧乙烷四氢呋喃共聚醚的数均分子量4000gmol
端三唑基聚环氧乙烷四氢呋喃共聚醚的制备方法为:
1)将100g端叠氮基聚环氧乙烷四氢呋喃共聚醚溶于200mL四氢呋喃溶剂中,置于配有机械搅拌、氮气保护的500mL三口烧瓶中,加入0.92g抗坏血酸钠、0.37g CuSO4,搅拌均匀;
2)将2.59g丙炔醇加入步骤1)的容器中;
3)将步骤2)的混合溶液在40℃、搅拌条件下反应4小时,冷却,除去氮气保护;
4)将步骤3)的反应混合物用蒸馏水萃取三次,旋蒸、干燥后得到96g端三唑基聚环氧乙烷四氢呋喃共聚醚。
产物红外分析表明,2100cm
列举实施方式中所用端羟基聚叠氮缩水甘油醚的数均分子量4100g mol
端三唑基聚叠氮缩水甘油醚的制备方法为:
1)将真空除水的端羟基聚叠氮缩水甘油醚25g、无水四氢呋喃溶剂20mL放入带有机械搅拌和冷凝管装置250mL三口瓶中;
2)室温下,将3.26g TDI加入步骤1)的容器中,搅拌均匀后升温至65℃反应6小时,然后降至室温;
3)往步骤2)容器中加入7.08g 1-苯甲基-4-羟甲基三唑,搅拌均匀后升温至65℃反应6小时;
4)旋蒸除去步骤3)中的四氢呋喃溶剂,得到端三唑基聚叠氮缩水甘油醚粗品;
5)将步骤4)所得端三唑基聚叠氮缩水甘油醚粗品溶于20mL DMF溶剂,加入20mL蒸馏水振荡、静置分层,取油相减压蒸馏后得到端三唑基聚叠氮缩水甘油醚。
产物红外分析表明,产物端三唑基聚叠氮缩水甘油醚在3500cm
列举实施方式中所用端羟基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚的数均分子量3800g mol
端三唑基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚的制备方法为:
1)将真空除水的端羟基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚25g、无水四氢呋喃溶剂20mL放入带有机械搅拌和冷凝管装置250mL三口瓶中;
2)室温下,将3.26g TDI加入步骤1)的容器中,搅拌均匀后升温至65℃反应6小时,然后降至室温;
3)往步骤2)容器中加入7.08g 1-苯甲基-4-羟甲基三唑,搅拌均匀后升温至65℃反应6小时;
4)旋蒸除去步骤3)中的四氢呋喃溶剂,得到端三唑基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚粗品;
5)将步骤4)所得端三唑基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚粗品溶于20mL DMF溶剂,加入20mL蒸馏水振荡、静置分层,取油相减压蒸馏后得到端三唑基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚。
产物红外分析表明,产物端三唑基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚在3500cm
列举实施例中所用高分子端三唑基聚丁二烯(TTPB)预聚物数均分子量3000gmol
实施例1
将20g端三唑基聚丁二烯放入干燥的烧杯中,加入三溴新戊醇固化剂1.452g,搅拌均匀。然后真空浇注至四氟乙烯模具中,90℃下固化6天,得到三唑鎓离子交联的聚丁二烯弹性体。该弹性体20℃下力学拉伸强度1.35MPa,延伸率180%。
将得到的弹性体1g浸泡于5g 1-溴代戊烷溶剂中,5天后弹性体完全溶解,变为液态。
实施例2
将20g端三唑基聚环氧乙烷四氢呋喃共聚醚放入干燥的烧杯中,加入三溴新戊醇固化剂1.012g,搅拌均匀。然后真空浇注至四氟乙烯模具中,90℃下固化6天,得到三唑鎓离子交联的聚环氧乙烷四氢呋喃共聚醚弹性体。该弹性体20℃下力学拉伸强度1.25MPa,延伸率130%。
将得到的弹性体1g浸泡于5g 1-溴代戊烷溶剂中,5天后弹性体完全溶解,变为液态。
实施例3
将20g端三唑基聚环氧乙烷四氢呋喃共聚醚放入干燥的烧杯中,加入1,6-二溴代己烷0.285g、三溴新戊醇0.759g混合固化剂,搅拌均匀。然后真空浇注至四氟乙烯模具中,90℃下固化6天,得到三唑鎓离子交联的聚环氧乙烷四氢呋喃共聚醚弹性体。该弹性体20℃下力学拉伸强度1.05MPa,延伸率150%。
将得到的弹性体1g浸泡于5g 1-溴代戊烷溶剂中,5天后弹性体完全溶解,变为液态。
实施例4
将20g端三唑基聚叠氮缩水甘油醚预聚物放入干燥的烧杯中,确保三唑基团摩尔数与溴原子摩尔数等当量,加入相应的三溴新戊醇固化剂0.997g,搅拌均匀。然后真空浇注至四氟乙烯模具中,90℃下固化6天,得到三唑鎓离子交联的聚叠氮缩水甘油醚弹性体。该弹性体20℃下力学拉伸强度1.35MPa,延伸率120%。
将得到的弹性体1g浸泡于5g 1-溴代戊烷溶剂中,5天后弹性体完全溶解,变为液态。
实施例5
将20g端三唑基聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚预聚物放入干燥的烧杯中,加入1,6-二碘己烷0.389g、三溴新戊醇0.748g混合固化剂,搅拌均匀。然后真空浇注至四氟乙烯模具中,90℃下固化6天,得到三唑鎓离子交联的聚3,3-二叠氮甲基氧丁环四氢呋喃共聚醚弹性体。该弹性体20℃下力学拉伸强度1.15MPa,延伸率150%。
将1g该弹性体浸泡于5g 1-碘代辛烷溶剂中,5天后弹性体完全溶解,变为液态。
综上所述,以上仅为本发明的较佳实施例而已,并非用于限定本发明的保护范围。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
一种可溶解的三维交联弹性体及其制备和处理方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0