








专利摘要
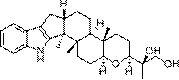
本发明公开了一类脱氢枞酸喹喔啉衍生物及其制备方法和应用,所述一种具有通式(Ⅰ)所示结构的脱氢枞酸喹喔啉衍生物I-a至I-i及其在药学上可接受的盐:,其中,脱氢枞酸喹喔啉衍生物I-a至I-i对应的R分别为:所述的通式I所示结构的脱氢枞酸喹喔啉衍生物I-a至I-i的制备方法,包括如下步骤:。本发明公开的化合物可以制成药学上可接受的盐或药学上可接受的载体。药理学实验表明,本发明的脱氢枞酸衍生物对肝癌细胞(HepG2)具有显著的抑制效果,具备作为抗肿瘤药物的开发价值。
权利要求
1.一种具有通式(Ⅰ)所示结构的脱氢枞酸喹喔啉衍生物I-a至I-i及其在药学上可接受的盐:
其中,脱氢枞酸喹喔啉衍生物I-a至I-i对应的R分别为:
2.权利要求1所述的通式I所示结构的脱氢枞酸喹喔啉衍生物I-a至I-i的制备方法,其特征在于包括如下步骤:
(1)a.12-溴-13,14-双氨基脱异丙基脱氢枞酸甲酯(II)溶于冰醋酸,加入1,2-二溴-2,3-丁二酮(II),在N2保护下,在120℃下,搅拌回流反应,合成得到12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯(III);
(2)b.12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯(III),加入催化剂碳酸钾,缚酸剂碘化钾,搅拌回流反应得到脱氢枞酸喹喔啉衍生物(I)。
3.如权利要求2所述的通式I所示结构的脱氢枞酸喹喔啉衍生物I-a至I-i的制备方法,其特征在于:
在步骤(1)中,称取12-溴-13,14-双氨基脱异丙基脱氢枞酸甲酯(II)溶于冰醋酸,加入1,2-二溴-2,3-丁二酮,所述12-溴-13,14-双氨基脱异丙基脱氢枞酸甲酯(II)与1,2-二溴-2,3-丁二酮的摩尔比为1:1,在120℃下,并在N2保护下,搅拌回流反应2小时;反应结束后,待反应液冷却到室温,用二氯甲烷萃取三次,合并有机层,依次用水、饱和碳酸氢钠溶液、饱和氯化钠溶液洗涤,无水硫酸钠除水,减压蒸馏除去有机溶剂,用硅胶柱对产物进行分离纯化,选用石油醚丙酮体系所述石油醚和丙酮的体积比500:1,得到黄色固体12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯(III);
在步骤(2)中,原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III溶于10mL的乙腈溶液中,一次加入碳酸钾,碘化钾和二甲胺,在85℃搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和二甲胺的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-a。
4.如权利要求3所述的通式I所示结构的脱氢枞酸喹喔啉衍生物I-a至I-i的制备方法,其特征在于:
将权利要求3中的步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和二乙胺,一次加入碳酸钾,碘化钾和二乙胺,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和二乙胺的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-b。
5.如权利要求3所述的通式I所示结构的脱氢枞酸喹喔啉衍生物I-a至I-i的制备方法,其特征在于:
将权利要求3中的步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和哌啶,一次加入碳酸钾,碘化钾和哌啶,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和哌啶的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用石油醚:丙酮体系,选用石油醚丙酮体系,所述石油醚和丙酮的体积比100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-c。
6.如权利要求3所述的通式I所示结构的脱氢枞酸喹喔啉衍生物I-a至I-i的制备方法,其特征在于:
将权利要求3中的步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物和吗啉,一次加入碳酸钾,碘化钾和吗啉,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和吗啉的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用石油醚:丙酮体系,所述石油醚和丙酮的体积比100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-d;
或者将权利要求3中的步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和N-甲基哌嗪,一次加入碳酸钾,碘化钾和N-甲基哌嗪,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和N-甲基哌嗪的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-e。
7.如权利要求3所述的通式I所示结构的脱氢枞酸喹喔啉衍生物I-a至I-i的制备方法,其特征在于:
将权利要求3中的步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和N-乙基哌嗪,一次加入碳酸钾,碘化钾和N-乙基哌嗪,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和N-乙基哌嗪的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-f;
或者将权利要求3中的步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和1,2,4-1H-三氮唑,一次加入碳酸钾,碘化钾和1,2,4-1H-三氮唑,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和1,2,4-1H-三氮唑的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-g。
8.如权利要求2所述的通式I所示结构的脱氢枞酸喹喔啉衍生物I-a至I-i的制备方法,其特征在于:
将权利要求3中的步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和四氮唑,一次加入碳酸钾,碘化钾和四氮唑,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和四氮唑的摩尔比为0.1:2:1:6;由于互变异构作用,会生成1-H四氮唑基和2-H四氮唑基两种取代产物;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,分别得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-h和脱氢枞酸喹喔啉衍生物I-i。
9.权利要求1所述的脱氢枞酸喹喔啉衍生物I-a至I-i及其药学上可接受的盐在制备治疗肿瘤药物中的应用。
10.根据权利要求9所述的应用,其特征在于:所述肿瘤为肝癌。
说明书
技术领域
本发明涉及天然产物有机合成及药物化学领域,具体涉及一类脱氢枞酸喹喔啉衍生物及其制备方法和应用。
背景技术
肿瘤是全世界死亡率最高的重大恶性疾病之一。肿瘤分为良性肿瘤和恶性肿瘤两大类,恶性肿瘤生长迅速,可转移到身体其他部位,破坏正常其关节欧股,使机体功能丧失,甚至死亡。肿瘤治疗的常规方法主要为化疗,即使用DNA合成抑制剂或细胞分裂抑制剂之类的细胞毒制剂来抑制肿瘤细胞,但同时,这些制剂也会杀伤正常的增殖较快的细胞,引起感染、出血等症状。因此,开发出选择性较、安全性好、疗效高的肿瘤抑制药物是现代肿瘤疾病研究的重要方向。
脱氢枞酸,是一种由歧化松香分离提纯得到的具有三环菲骨架的二萜类一元树脂酸。脱氢枞酸分子骨架上不存在容易氧化的双键,而含有苯环,因此性质稳定,适宜作为生物活性衍生物的原料。脱氢枞酸衍生物具有多种生物活性,如抗菌、抗肿瘤、抗病毒活性等。特别是脱氢枞酸衍生物在抗肿瘤方面的显著活性,已引起国内外研究者的注意。越来越多的报道已表明脱氢枞酸作为一种前体化合物,在抗肿瘤方面的研究具有较大的价值。
喹喔啉衍生物作为一类重要的苯并吡嗪类杂环化合物,是一种重要的化工中间体,在动物促生长剂、抗细菌、抗肿瘤细胞、抗结核杆菌、抗寄生虫、荧光探针及染料中间体等领域具有较大的潜在应用价值。很多研究结果表明,含喹喔啉结构的化合物具有一定的药理活性,可应用为抗肿瘤方面的制剂。
其中,化合物II根据文献(T.Fenseca,B.Gigante,M.M.Marques,et al.Bioorg.Med.Chem.Lett.2004,12,103-112)报道方法合成。在该文献中报道了三个脱氢枞酸喹喔啉衍生物,其喹喔啉环上分别有两个甲基、丁基或苯基取代基。这三个化合物具有一定的抗病毒活性,但细胞毒活性很弱。因此如要开发抗肿瘤药物,还需对其结构进行改造。许多研究表明,在药物分子中引入一些含氮基团,例如脂肪胺基、脂肪族含氮基团、唑类芳杂环可以改变化合物的极性与酸碱性,提高化合物的溶解性,通过氢键或芳环π堆积作用增强药物分子与细胞中生物大分子靶点的结合能力,因而可以有效的提高化合物的抗肿瘤活性(K.R.Wang,Y.Q.Wang,X.H.Yan,et al.Bioorg.Med.Chem.Lett.2012,22,937-941;J.S.Lv,X.M.Peng,B.Kishore,et al.Bioorg.Med.Chem.Lett.2014,24,308-313;M.A.Islam,Y.Q.Zhang,Y.Wang,et al.Med.Chem.Commun.2015,6,300-305)。
因此,为了更深入研究脱氢枞酸杂环衍生物的构效关系,寻找具有较高抗肿瘤活性的药物先导物,本发明提供一类具有抗肿瘤活性的新型脱氢枞酸喹喔啉衍生物,该类衍生物是对脱氢枞酸苯环进行衍生化,骈合喹喔啉杂环。并喹喔啉杂环上进一步引入不同的脂肪族和芳香族含氮基团。此类化合物的合成及抗肿瘤活性的研究国内外均未见报道。另外还对此类化合物在抗肿瘤方面的活性进行了研究。
发明内容
为了克服上述现有技术中的不足之处,本发明的目的是提供一种抗肿瘤活性较高的脱氢枞酸喹喔啉衍生物及其制备方法和应用。
为了实现上述技术目的,本发明采用的技术方案的如下:本发明的一种具有通式(Ⅰ)所示结构的脱氢枞酸喹喔啉衍生物I-a至I-i及其在药学上可接受的盐:
其中,脱氢枞酸喹喔啉衍生物I-a至I-i对应的R分别为:
本发明所述的通式I所示结构的脱氢枞酸喹喔啉衍生物I-a至I-i的制备方法,包括如下步骤:
(1)a.12-溴-13,14-双氨基脱异丙基脱氢枞酸甲酯(II)溶于冰醋酸,加入1,2-二溴-2,3-丁二酮(II),在N2保护下,在120℃下,搅拌回流反应,合成得到12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯(III);
(2)b.12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯(III),加入催化剂碳酸钾,缚酸剂碘化钾,搅拌回流反应得到脱氢枞酸喹喔啉衍生物(I)。
进一步地,在步骤(1)中,称取12-溴-13,14-双氨基脱异丙基脱氢枞酸甲酯(II)溶于冰醋酸,加入1,2-二溴-2,3-丁二酮,所述12-溴-13,14-双氨基脱异丙基脱氢枞酸甲酯(II)与1,2-二溴-2,3-丁二酮的摩尔比为1:1,在120℃下,并在N2保护下,搅拌回流反应2小时;反应结束后,待反应液冷却到室温,用二氯甲烷萃取三次,合并有机层,依次用水、饱和碳酸氢钠溶液、饱和氯化钠溶液洗涤,无水硫酸钠除水,减压蒸馏除去有机溶剂,用硅胶柱对产物进行分离纯化,选用石油醚丙酮体系所述石油醚和丙酮的体积比500:1,得到黄色固体12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯(III);
在步骤(2)中,原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III溶于10mL的乙腈溶液中,一次加入碳酸钾,碘化钾和二甲胺,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和二甲胺的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-a。
进一步地,将步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和二乙胺,一次加入碳酸钾,碘化钾和二乙胺,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和二乙胺的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-b。
更进一步地,将步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和哌啶,一次加入碳酸钾,碘化钾和哌啶,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和哌啶的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用石油醚:丙酮体系,选用石油醚丙酮体系,所述石油醚和丙酮的体积比100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-c。
进一步地,将步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物和吗啉,一次加入碳酸钾,碘化钾和吗啉,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和吗啉的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用石油醚:丙酮体系,所述石油醚和丙酮的体积比100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-d;
或者将步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和N-甲基哌嗪,一次加入碳酸钾,碘化钾和N-甲基哌嗪,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和N-甲基哌嗪的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-e。
进一步地,将步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和N-乙基哌嗪,一次加入碳酸钾,碘化钾和N-乙基哌嗪,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和N-乙基哌嗪的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-f;
或者将步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和1,2,4-1H-三氮唑,一次加入碳酸钾,碘化钾和1,2,4-1H-三氮唑,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和1,2,4-1H-三氮唑的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-g。
更进一步地,将步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和四氮唑,一次加入碳酸钾,碘化钾和四氮唑,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和四氮唑的摩尔比为0.1:2:1:6;由于互变异构作用,会生成1-H四氮唑基和2-H四氮唑基两种取代产物;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,分别得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-h和脱氢枞酸喹喔啉衍生物I-i。
本发明所述的脱氢枞酸喹喔啉衍生物I-a至I-i及其药学上可接受的盐在制备治疗肿瘤药物中的应用。
进一步地,所述肿瘤为肝癌。
有益效果:本发明本发明公开的化合物可以制成药学上可接受的盐或药学上可接受的载体。药理学实验表明,本发明的脱氢枞酸衍生物对肝癌细胞(HepG2)具有显著的抑制效果,具备作为抗肿瘤药物的开发价值。本发明对于天然产物脱氢枞酸进行结构修饰,得到一系列新型脱氢枞酸喹喔啉衍生物。通过体外细胞增殖实验表明,本系列化合物对肿瘤细胞(HepG2)具有良好的抑制作用。本发明公开的化合物可制成药学上可接受的盐或可接受的载体。本发明的药学上可接受的盐与其化合物具有同样的功效。
具体实施方式
以下通过实施例进一步说明本发明。应该理解的是,这些实施例是本发明的阐释和举例,并不以任何形式限制本发明的范围。
本发明的一种具有通式(Ⅰ)所示结构的脱氢枞酸喹喔啉衍生物I-a至I-i及其在药学上可接受的盐:
其中,脱氢枞酸喹喔啉衍生物I-a至I-i对应的R分别为:
本发明所述的通式I所示结构的脱氢枞酸喹喔啉衍生物I-a至I-i的制备方法,包括如下步骤:
(1)a.12-溴-13,14-双氨基脱异丙基脱氢枞酸甲酯(II)溶于冰醋酸,加入1,2-二溴-2,3-丁二酮(II),在N2保护下,在120℃下,搅拌回流反应,合成得到12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯(III);
(2)b.12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯(III),加入催化剂碳酸钾,缚酸剂碘化钾,搅拌回流反应得到脱氢枞酸喹喔啉衍生物(I)。
在步骤(1)中,称取12-溴-13,14-双氨基脱异丙基脱氢枞酸甲酯(II)溶于冰醋酸,加入1,2-二溴-2,3-丁二酮,所述12-溴-13,14-双氨基脱异丙基脱氢枞酸甲酯(II)与1,2-二溴-2,3-丁二酮的摩尔比为1:1,在120℃下,并在N2保护下,搅拌回流反应2小时;反应结束后,待反应液冷却到室温,用二氯甲烷萃取三次,合并有机层,依次用水、饱和碳酸氢钠溶液、饱和氯化钠溶液洗涤,无水硫酸钠除水,减压蒸馏除去有机溶剂,用硅胶柱对产物进行分离纯化,选用石油醚丙酮体系所述石油醚和丙酮的体积比500:1,得到黄色固体12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯(III);
在步骤(2)中,原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III溶于10mL的乙腈溶液中,一次加入碳酸钾,碘化钾和二甲胺,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和二甲胺的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-a。
将步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和二乙胺,一次加入碳酸钾,碘化钾和二乙胺,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和二乙胺的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-b。
将步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和哌啶,一次加入碳酸钾,碘化钾和哌啶,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和哌啶的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用石油醚:丙酮体系,选用石油醚丙酮体系,所述石油醚和丙酮的体积比100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-c。
将步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物和吗啉,一次加入碳酸钾,碘化钾和吗啉,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和吗啉的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用石油醚:丙酮体系,所述石油醚和丙酮的体积比100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-d;
或者步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和N-甲基哌嗪,一次加入碳酸钾,碘化钾和N-甲基哌嗪,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和N-甲基哌嗪的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-e。
将步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和N-乙基哌嗪,一次加入碳酸钾,碘化钾和N-乙基哌嗪,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和N-乙基哌嗪的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-f;
或者步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和1,2,4-1H-三氮唑,一次加入碳酸钾,碘化钾和1,2,4-1H-三氮唑,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和1,2,4-1H-三氮唑的摩尔比为0.1:2:1:6;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-g。
将步骤(2)替换为:原料为12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III和四氮唑,一次加入碳酸钾,碘化钾和四氮唑,搅拌回流反应8h,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯、碳酸钾、碘化钾和四氮唑的摩尔比为0.1:2:1:6;由于互变异构作用,会生成1-H四氮唑基和2-H四氮唑基两种取代产物;
以乙腈作为溶剂,所述12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯化合物III的摩尔浓度为0.01mol/L,所述反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取;合并有机相,用水洗涤,再用碳酸氢钠饱和溶液洗涤,最后用饱和碳酸钠溶液洗涤,水硫酸钠干燥,蒸馏去除溶剂;
用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系,所述二氯甲烷与甲醇的体积比为100:1,分别得到淡黄色油状物脱氢枞酸喹喔啉衍生物I-h和脱氢枞酸喹喔啉衍生物I-i。
本发明所述的脱氢枞酸喹喔啉衍生物I-a至I-i及其药学上可接受的盐在制备治疗肿瘤药物中的应用。
所述肿瘤为肝癌。
实施例1
12-溴-13,14-二溴甲基喹喔啉环脱氢枞酸甲酯(III)的合成
称取12-溴-13,14-双氨基脱异丙基脱氢枞酸甲酯(II)0.130g(0.34mmol)溶于冰醋酸,加入的1,2-二溴-2,3-丁二酮0.083g(0.34mmol),在120℃下,并在N2保护下,搅拌回流反应2小时。反应结束后,待反应液冷却到室温,用二氯甲烷萃取三次,合并有机层,依次用水、饱和碳酸氢钠溶液、饱和氯化钠溶液洗涤,无水硫酸钠除水,减压蒸馏除去有机溶剂。用硅胶柱对产物进行分离纯化,选用石油醚丙酮体系(体积比500:1),得到黄色固体III 0.118g,得率为90.8%。本发明采用的纯化方法有:重结晶与正相硅胶柱层析分离纯化方法,步骤如下:
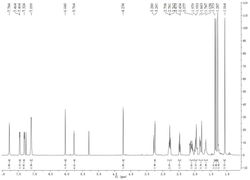
1H NMR(600MHz,CDCl3):δ1.30(s,3H),1.32(s,3H),1.50~1.90(m,7H),2.30(dd,J=12.6,1.8Hz,1H),2.38(d,J=13.0Hz,1H),3.18(ddd,J=19.1,11.5,7.7Hz,1H),3.52(dd,J=18.8,6.1Hz,1H),3.70(s,3H,COOCH3),4.94(s,2H),4.97(s,2H),8.02(s,1H).IR(KBr,cm-1):2926,2853,1726,1588,1540,1457,1248,1186,1028,960.HRMS(ESI):m/z[M+H]+calcd for C22H26Br3N2O2:586.9544;found:586.9552.
脱氢枞酸喹喔啉衍生物I-a的合成
原料为化合物III 0.0589g(0.1mmol)溶于10mL的乙腈溶液中,一次加入碳酸钾0.2764g(2mmol),碘化钾0.166g(1mmol)和二甲胺0.2705g(6mmol),搅拌回流反应8h。反应完成后待反应物冷却到室温,将反应物倒入水中,用二氯甲烷萃取3次,每次50mL。合并有机相,用水洗涤3次,每次50mL,再用50mL碳酸氢钠饱和溶液洗涤一次,最后用50mL饱和碳酸钠溶液洗涤一次,水硫酸钠干燥,蒸馏去除溶剂。用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系(100:1),得到淡黄色油状物I-a 0.0269g,得率为52.0%。将反应产物溶解后与等量柱层析硅胶(300目~400目)混合拌样,挥干溶剂;将适当倍数的硅胶采用湿法装柱的方法装入层析柱中,将吸附了样品的硅胶装入柱中;采用一定比例的洗脱剂依次洗脱,用薄层层析分析洗脱下来的成分,合并相同成分,减压蒸馏去除溶剂得到目标产物。
1H NMR(300MHz,CDCl3):δ1.31(s,3H),1.33(s,3H),1.50~1.90(m,7H),2.10~2.40(m,2H),2.40(s,6H),2.47(s,6H),3.20(m,1H),3.56(dd,J=18.4,6.0Hz,1H),3.69(s,3H),4.07(brs,2H),4.14(brs,2H),7.96(s,1H).IR(KBr,cm-1):2924,2854,1726,1616,1464,1383,1248,1124,1034,988;HRMS(ESI):m/z[M+H]+calcd for C26H38BrN4O2:517.2178;found:517.2182.
实施例2
脱氢枞酸喹喔啉衍生物I-b的合成
参照实施例1的方法,原料为化合物III 0.0589g(0.1mmol)和二乙胺0.4388g(6mmol),在相同条件下反应8h,用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系(100:1),得到淡黄色油状物I-b 0.0189g,得率为32.9%。
1H NMR(300MHz,CDCl3):δ1.07(t,J=7.0Hz,6H),1.11(t,J=7.1Hz,6H),1.30(s,3H),1.33(s,3H),1.50~1.90(m,7H),2.10~2.40(m,2H),2.75(q,J=7.1Hz,4H),2.87(q,J=6.8Hz,4H),3.15(m,1H),3.53(dd,J=19.0,5.9Hz,1H),3.70(s,3H),4.22(brs,2H),4.31(brs,2H),7.95(s,1H).IR(KBr,cm-1):2926,2854,1726,1613,1462,1384,1247,1186,1125,1081,966;HRMS(ESI):m/z[M+H]+calcd for C30H46BrN4O2:573.2804;found:573.2809.
实施例3
脱氢枞酸喹喔啉衍生物I-c的合成
参照实施例1的方法,原料为化合物III 0.0589g(0.1mmol)和哌啶0.5109g(6mmol),在相同条件下反应8h,用硅胶柱对产物进行分离纯化,选用石油醚:丙酮体系(100:1),得到淡黄色油状物I-c 0.0284g,得率为47.5%。
1H NMR(600MHz,CDCl3):δ1.29(s,3H),1.32(s,3H),1.40~1.47(m,4H),1.50~1.56(m,9H),1.61(dd,J=13.0,7.9Hz,1H),1.68(dd,J=9.5,3.4Hz,1H),1.75~1.90(m,4H),2.30(dd,J=12.6,1.9Hz,1H),2.37(d,J=12.8Hz,1H),2.47(brs,4H),2.50(brs,4H),3.16(ddd,J=19.0,11.2,7.8Hz,1H),3.54(dd,J=18.9,6.0Hz,1H),3.69(s,3H,COOCH3),4.03(d,J=13.0Hz,1H),4.05(d,J=14.0Hz,1H),4.07(d,J=11.4Hz,1H),4.11(d,J=13.1Hz,1H),7.92(s,1H).IR(KBr,cm-1):2931,2853,1728,1591,1463,1383,1247,1125,986;HRMS(ESI):m/z[M+H]+calcd for C32H46BrN4O2:597.2804;found:597.2811.
实施例4
脱氢枞酸喹喔啉衍生物I-d的合成
参照实施例1的方法,原料为化合物III 0.0589g(0.1mmol)和吗啉0.5227g(6mmol),在相同条件下反应8h,用硅胶柱对产物进行分离纯化,选用石油醚:丙酮体系(100:1),得到淡黄色油状物I-d 0.0378g,得率为62.8%。
1H NMR(600MHz,CDCl3):δ1.30(s,3H),1.32(s,3H),1.54(dt,J=12.6,3.1Hz,1H),1.62(dd,J=12.8,8.0Hz,1H),1.70(dd,J=12.7,3.7Hz,1H),1.75~1.90(m,4H),2.30(dd,J=12.5,1.8Hz,1H),2.38(d,J=12.9Hz,1H),2.55(m,4H),2.60(m,4H),3.16(ddd,J=18.9,11.2,7.4Hz,1H),3.53(dd,J=18.7,6.1Hz,1H),3.68(m,8H),3.69(s,3H,COOCH3),4.05(d,J=13.0Hz,1H),4.10(d,J=13.0Hz,1H),4.11(d,J=13.3Hz,1H),4.15(d,J=13.1Hz,1H),7.95(s,1H).IR(KBr,cm-1):2926,2852,1727,1615,1453,1382,1324,1247,1119,1007,865;HRMS(ESI):m/z[M+H]+calcd for C30H42BrN4O4:601.2389;found:601.2383.
实施例5
脱氢枞酸喹喔啉衍生物I-e的合成
参照实施例1的方法,原料为化合物III 0.0589g(0.1mmol)和N-甲基哌嗪0.6010g(6mmol),在相同条件下反应8h,用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系(100:1),得到淡黄色油状物I-e 0.0245g,得率为39.0%。
1H NMR(300MHz,CDCl3):δ1.30(s,3H),1.33(s,3H),1.50~1.90(m,7H),2.10~2.40(m,2H),2.31(s,3H),2.32(s,3H),2.48(brs,8H),2.58(brs,4H),2.65(brs,4H),3.17(ddd,J=18.8,11.0,7.5Hz,1H),3.55(dd,J=18.7,6.2Hz,1H),3.70(s,3H),4.08(brs,2H),4.13(brs,2H),7.95(s,1H).IR(KBr,cm-1):2930,2849,2796,1727,1592,1456,1372,1247,1162,1010,814;HRMS(ESI):m/z[M+H]+calcd for C32H48BrN6O2:627.3022;found:627.3028.
实施例6
脱氢枞酸喹喔啉衍生物I-f的合成
参照实施例1的方法,原料为化合物III 0.0589g(0.1mmol)和N-乙基哌嗪0.6851g(6mmol),在相同条件下反应8h,用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系(100:1),得到淡黄色油状物I-f 0.0195g,得率为29.7%。
1H NMR(300MHz,CDCl3):δ1.11(t,J=7.0Hz,3H),1.13(t,J=7.1Hz,3H),1.29(s,3H),1.32(s,3H),1.50~1.90(m,7H),2.10~2.40(m,2H),2.48(m,8H),2.51(m,4H),2.63(brs,4H),2.71(brs,4H),3.15(ddd,J=18.8,11.2,8.0Hz,1H),3.53(dd,J=18.8,6.0Hz,1H),3.69(s,3H),4.06(brs,2H),4.13(brs,2H),7.94(s,1H).IR(KBr,cm-1):2926,2852,2812,1728,1667,1594,1462,1380,1246,1125,966;HRMS(ESI):m/z[M+H]+calcd for C34H51BrN6O2:654.3257;found:654.3250.
实施例7
脱氢枞酸喹喔啉衍生物I-g的合成
参照实施例1的方法,原料为化合物III 0.0589g(0.1mmol)和1,2,4-1H-三氮唑0.4144g(6mmol),在相同条件下反应8h,用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系(100:1),得到淡黄色油状物I-g 0.0259g,得率为45.8%。
1H NMR(300MHz,CDCl3):δ1.28(s,3H),1.31(s,3H),1.50~1.90(m,7H),2.24(d,J=11.2Hz,1H),2.34(d,J=12.7Hz,1H),2.97(m,1H),3.23(dd,J=18.8,6.0Hz,1H),3.70(s,3H),5.79(s,2H),5.87(s,2H),7.98(s,1H),8.41(brs,2H),8.54(brs,2H).IR(KBr,cm-1):2924,2854,1720,1594,1462,1363,1249,1188,1080,969;HRMS(ESI):m/z[M+H]+calcd for C26H30BrN8O2:565.1675;found:565.1681.
实施例8
脱氢枞酸喹喔啉衍生物I-h和I-i的合成
参照实施例1的方法,原料为化合物III 0.0589g(0.1mmol)和四氮唑0.4203g(6mmol),在相同条件下反应8h。由于互变异构作用,会生成1-H四氮唑基和2-H四氮唑基两种取代产物。用硅胶柱对产物进行分离纯化,选用二氯甲烷:甲醇体系(100:1),得到淡黄色油状物I-h 0.0201g,I-i 0.0172g,得率分别为35.4%,30.3%。
化合物I-h:1H NMR(300MHz,CDCl3):δ1.26(s,3H),1.30(s,3H),1.50~1.90(m,7H),2.23(d,J=11.1Hz,1H),2.34(d,J=12.6Hz,1H),2.98(ddd,J=18.6,11.0,7.8Hz,1H),3.23(dd,J=19.2,6.3Hz,1H),3.70(s,3H),6.20(s,2H),6.22(s,2H),8.07(s,1H),8.94(s,1H),9.13(s,1H).IR(KBr,cm-1):2924,2872,1720,1663,1593,1470,1386,1248,1124,1023,958;HRMS(ESI):m/z[M+H]+calcd for C24H28BrN10O2:567.1580;found:567.1584.
化合物I-i:1H NMR(300MHz,CDCl3):δ1.26(s,3H),1.30(s,3H),1.50~1.90(m,7H),2.24(dd,J=12.3,5.1Hz,1H),2.35(d,J=12.3Hz,1H),2.98(ddd,J=18.7,11.3,7.9Hz,1H),3.34(dd,J=19.2,6.6Hz,1H),3.71(s,3H),6.41(s,2H),6.42(s,2H),8.06(s,1H),8.60(brs,2H).IR(KBr,cm-1):2925,2849,1717,1669,1466,1433,1385,1253,1169,1099,964;HRMS(ESI):m/z[M+H]+calcd for C24H28BrN10O2:567.1580;found:567.1575.
实施例9
体外抗肿瘤活性筛选
筛选细胞为人体肝癌细胞(HepG2)
实验方法:取对数生长期状态良好的细胞,经胰蛋白酶消化后,计算出细胞浓度,然后配置成4×104细胞/mL的细胞悬液。将细胞悬液用移液枪移入96孔板中,每孔200μL,将孔板置于37℃、5%CO2条件下培养24h。
将受试化合物用DMSO配置成2000μg/mL浓度的母液,再用磷酸缓冲液将衍生物母液稀释成不同作用浓度的稀释液,浓度分别为0.01μg/mL,0.02μg/mL,0.1μg/mL,0.2μg/mL,1μg/mL,2μg/mL,10μg/mL,20μg/mL。用移液枪移去孔内的培养基,按浓度梯度,分别加入不同浓度的药液,每孔200μg/mL,并做三次平行试验。另设空白对照组与阳性对照依托泊苷(VP-16)对照组。药物作用24h后,在每孔内加入浓度为5mg/mL的噻唑蓝溶液(MTT)20μg/mL,对细胞进行染色。操作完毕后,将孔板放入培养箱培养4h。
倒出孔板内的溶液,每孔分别加入150μg/mL DMSO溶液,振荡10分钟后测定溶液的吸光度值,其中酶联仪的波长λ设为490纳米。利用测得的各孔光吸收值(OD值),计算细胞的增值抑制率:
I(%)=(1-OD1/OD)×100%,其中,I=肿瘤细胞的抑制率;OD1=加药组肿瘤细胞吸光度值(平均值),OD=空白对照组肿瘤细胞吸光度值(平均值)。
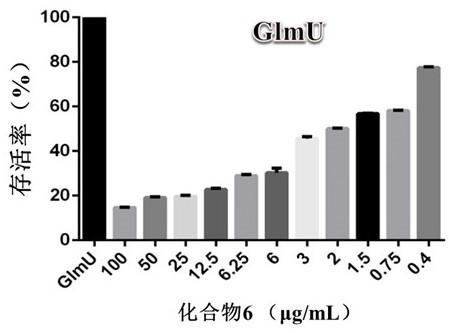
应用SPSS16.0软件进行数据处理并计算癌细胞增值的半数抑制浓度(IC50),表1为脱氢枞酸喹喔啉衍生物对人体肝癌细胞(HepG2)的体外增殖抑制作用。结果如表1所示,
表1
如表1结果所示,这一系列的脱氢枞酸喹喔啉衍生物对人体肝癌细胞(HepG2)具有较高的增殖抑制效果,显著强于其合成的前体化合物II和III。化合物II仅表现出中等活性(43.72μM),而化合物III在<50μM的范围内未表现出细胞毒活性。说明这些脂肪族或芳香族含氮基团的引入对于此类脱氢枞酸喹喔啉衍生物的抗肿瘤活性具有不可或缺的作用。在该系列化合物中,化合物I-a、I-b和I-g的IC50值分别为2.09μM、1.18μM和2.18μM,接近阳性对照依托泊苷。说明此类脱氢枞酸喹喔啉衍生物表现出较好的抗肿瘤作用,具有开发抗癌药物的潜力。
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,本发明要求保护范围由所附的权利要求书、说明书及其等效物界定。
一类脱氢枞酸喹喔啉衍生物及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0