

IPC分类号 : C07F5/00,C09K11/06,H01F1/42








专利摘要
本发明公开了两个8‑羟基喹啉衍生物和1,10‑邻菲啰啉混合掺杂的镝配合物及其制备方法和应用。这两个镝配合物为配合物1或配合物2,其中配合物1的化学式为:DyⅢ(L1)2(L2)(NO3),配合物2的化学式为:DyⅢ(L3)2(L2)(NO3),其中L1为2‑甲基‑5,7‑二溴‑8‑羟基喹啉脱去羟基氢原子,带一个单位负电荷;L2为1,10‑邻菲啰啉,L3为2‑甲基‑5,7‑二氯‑8‑羟基喹啉脱去羟基氢原子,带一个单位负电荷。两个配合和物均属于单斜晶系,P21/c空间群。本发明提供的两个配合物制备方法简单、产率较高、重复性好,且均具有场诱导的单分子磁体行为,可以用于制备磁性材料。
权利要求
1.8-羟基喹啉衍生物和1,10-林菲罗琳混合掺杂的镝配合物,为配合物1或配合物2,其中:
配合物1的化学式为:Dy
该配合物属于单斜晶系,P2
配合物2的化学式为:Dy
该配合物属于单斜晶系,P2
2.权利要求1所述的8-羟基喹啉衍生物和1,10-林菲罗琳混合掺杂的镝配合物的制备方法,其特征在于:
配合物1的制备方法为:将2-甲基-5,7-二溴-8-羟基喹啉、1,10-林菲罗琳以及Dy(NO
配合物2的制备方法为:将2-甲基-5,7-二氯-8-羟基喹啉、1,10-林菲罗琳以及Dy(NO
3.根据权利要求2所述的制备方法,其特征在于:所述第一混合溶剂的组成中,1,4二氧六环和水和体积比为1:2-4。
4.根据权利要求2所述的制备方法,其特征在于:所述第二混合溶剂的组成中,甲醇和乙腈的体积比为1:1-3。
5.根据权利要求2-4中任一项所述的制备方法,其特征在于:反应在≥50℃的条件下进行。
6.根据权利要求2-4中任一项所述的制备方法,其特征在于:反应在60-120℃的条件下进行。
7.根据权利要求2-4中任一项所述的制备方法,其特征在于:在配合物1的制备方法中,调节所得溶液的pH=5.5-5.6。
8.根据权利要求2-4中任一项所述的制备方法,其特征在于:在配合物2的制备方法中,调节所得溶液的pH=4.1-4.2。
9.权利要求1所述的8-羟基喹啉衍生物和1,10-林菲罗琳混合掺杂的镝配合物在制备磁性材料中的应用。
说明书
技术领域
本发明涉及8-羟基喹啉衍生物和1,10-林菲罗琳混合掺杂的镝配合物及其制备方法和应用,属于磁性材料技术领域。
背景技术
近十多年来,人们对基于镧系元素的配位化合物的兴趣日益增长,特别是它们有趣的光学、磁学性质。将两种性质结合到一个分子实体中是提供双功能分子的最有效途径。镧系元素作为发光单分子磁体(SMMs)构建的一组优选目标,由于其在SMM领域的优异性能和分子材料的发光性,使其广泛用于高密度数据存储、量子计算、发光二极管和彩色显示器中的背光源,以及生物医学成像,光动力疗法和其他光子器件。由于4fn电子配置的多个电子层,Ln(III)离子显示出特定的发射,大部分在光谱范围从可见光区域到红外(NIR)区域。
基于镧系元素的多色发光材料的设计通常有多种,但目前尚未见有将8- 羟基喹啉衍生物和1,10-林菲罗琳同时引入到镝配合物中以实现其可调发射型发光的相关报道。
发明内容
本发明要解决的技术问题是提供两个具有单分子磁体行为的可调发射型发光的8-羟基喹啉衍生物和1,10-林菲罗琳混合掺杂的镝配合物及其制备方法和应用。
本发明所述的8-羟基喹啉衍生物和1,10-林菲罗琳混合掺杂的镝配合物,为配合物1或配合物2,其中:
配合物1的化学式为:Dy
该配合物属于单斜晶系,P21/c空间群,晶胞参数为: α=90.00°,β=97.666(4)°,γ=90.00°;
配合物2的化学式为:Dy
该配合物属于单斜晶系,P21/c空间群,晶胞参数为: α=90.00°,β=97.7270(10)°,γ=90.00°。
本发明还提供上述8-羟基喹啉衍生物和1,10-林菲罗琳混合掺杂的镝配合物的制备方法,具体的:
配合物1的制备方法为:将2-甲基-5,7-二溴-8-羟基喹啉、1,10-林菲罗琳以及Dy(NO3)3·6H2O置于第一混合溶剂中溶解,调节所得溶液的pH=5.5-6.0,所得混合液于加热条件下反应,即得;其中,所述的第一混合溶剂为1,4二氧六环和水的组合物;
配合物2的制备方法为:将2-甲基-5,7-二氯-8-羟基喹啉、1,10-林菲罗琳以及Dy(NO3)3·6H2O置于第二混合溶剂中溶解,调节所得溶液的pH=4.0-4.5,所得混合液于加热条件下反应,即得;其中,所述的第二混合溶剂为甲醇和乙腈的组合物。
上述配合物1的制备方法中,2-甲基-5,7-二溴-8-羟基喹啉、1,10-林菲罗琳和Dy(NO3)3·6H2O的摩尔比为化学计量比,在实际操作过程中, Dy(NO3)3·6H2O的量可相对过量一些。可以采用现有常用的碱性物质(如氨水、三乙胺、碳酸氢钠、碳酸钠、碳酸钾等)来调节溶液的pH值,优选是采用三乙胺调节溶液的pH值;更优选是采用三乙胺调节所得溶液的 pH=5.5-5.6。在第一混合溶剂的组成中,1,4二氧六环和水和体积比优选为1: 2-4,优选为1:2.5-3.5。所述混合溶剂的用量可根据需要确定,通常以能溶解参加反应的原料为宜。具体地,以1mmol的2-甲基-5,7-二溴-8-羟基喹啉为基准计算,全部原料所用混合溶剂的总用量一般为14-18mL。在具体溶解的步骤中,可将各原料分别用混合溶剂中的某一种组分溶解,然后再混合在一起反应;也可将所有原料混合在一起后再加混合溶剂溶解。
上述配合物2的制备方法中,2-甲基-5,7-二氯-8-羟基喹啉、1,10-林菲罗琳和Dy(NO3)3·6H2O的摩尔比为化学计量比,在实际操作过程中, Dy(NO3)3·6H2O的量可相对过量一些。可以采用现有常用的碱性物质(如氨水、三乙胺、碳酸氢钠、碳酸钠、碳酸钾等)来调节溶液的pH值,优选是采用三乙胺调节溶液的pH值;更优选是采用三乙胺调节所得溶液的 pH=4.1-4.2。在第二混合溶剂的组成中,甲醇和乙腈的体积比为2:1-2:2所述混合溶剂的用量可根据需要确定,通常以能溶解参加反应的原料为宜。具体地,以1mmol的2-甲基-5,7-二氯-8-羟基喹啉为基准计算,全部原料所用混合溶剂的总用量一般为15-20mL。在具体溶解的步骤中,可将各原料分别用混合溶剂中的某一种组分溶解,然后再混合在一起反应;也可将所有原料混合在一起后再加混合溶剂溶解。
在本发明所述的配合物1和配合物2的制备方法中,反应优选是在≥50℃的条件下进行,在上述温度条件下进行反应的时间通常为48-72h。反应更优选是在60-120℃条件下进行。
本发明所述的配合物1和配合物2的制备方法中,在上述优选的溶剂组成、pH值以及反应温度条件下,可以获得更高的产率,且所得晶体的质量更好。
申请人对本发明所述的8-羟基喹啉衍生物和1,10-林菲罗琳混合掺杂的镝配合物的磁性研究发现,该簇配合物的磁学性质表现为典型的单分子磁体行为。因此,本发明还包括上述镝簇合物在制备磁性材料中的应用。
与现有技术相比,本发明提供了两个结构新颖的8-羟基喹啉衍生物和 1,10-林菲罗琳混合掺杂的镝配合物及其制备方法,申请人还发现它们为具有单分子磁体行为的可调发射型发光镝配合物,可以用于制备磁性材料;此外,该镝合物的制备方法简单、成本低廉、重复性好。
附图说明
图1为本发明实施例1制得的最终产物的化学结构图;
图2为本发明实施例1制得的最终产物的热重曲线图;
图3为本发明实施例1制得的最终产物的粉末衍射图;
图4为本发明实施例5制得的最终产物的化学结构图;
图5为本发明实施例5制得的最终产物的热重曲线图;
图6为本发明实施例5制得的最终产物的粉末衍射图;
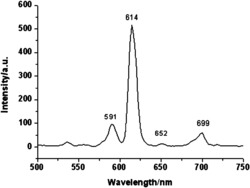
图7为本发明实施例1制得的最终产物的荧光光谱图;
图8为本发明实施例5制得的最终产物的荧光光谱图;
图9为本发明实施例1制得的最终产物在零直流中不同频率的实部(χ') 和虚部(χ”)交流磁化率对温度曲线图;
图10为本发明实施例5制得的最终产物在零直流中不同频率的实部(χ') 和虚部(χ”)交流磁化率对温度曲线图;
图11为本发明实施例1制得的最终产物(即配合物1)和本发明实施例5 制得的最终产物(即配合物2)在0Oe场条件下在不同温度下的频率依赖的交流磁化率曲线以及加0Oe场下从交流磁化率中得出的cole-cole曲线,其中, (a)和(b)为配合物2在0Oe场条件下在不同温度下的频率依赖的交流磁化率曲线,(c)对于配合物2在加0Oe场下从交流磁化率中得出的cole-cole曲线,(d) 和(e)为配合物1在0Oe场条件下在不同温度下的频率依赖的交流磁化率曲线,(f)对于配合物1在加0Oe场下从交流磁化率中得出的cole-cole曲线;
图12为本发明实施例1制得的最终产物(即配合物1)和本发明实施例5 制得的最终产物(即配合物2)在1500Oe直流磁场中不同频率的实部(χ')和虚部(χ”)交流磁化率对温度曲线图,其中,(a)为配合物2在1500Oe直流磁场中不同频率的实部(χ')和虚部(χ”)交流磁化率对温度曲线,(b)为配合物 1在1500Oe直流磁场中不同频率的实部(χ')和虚部(χ”)交流磁化率对温度曲线;
图13为本发明实施例1制得的最终产物(即配合物1)和本发明实施例5 制得的最终产物(即配合物2)在1500Oe场条件下在不同温度下的频率依赖的交流磁化率曲线以及加1500Oe场下从交流磁化率中得出的cole-cole曲线,其中,(a)和(b)为配合物2在1500Oe场条件下在不同温度下的频率依赖的交流磁化率曲线,(c)对于配合物2在加1500Oe场下从交流磁化率中得出的 cole-cole曲线,(d)和(e)为配合物1在1500Oe场条件下在不同温度下的频率依赖的交流磁化率曲线,(f)对于配合物1在加1500Oe场下从交流磁化率中得出的cole-cole曲线;
图14为本发明实施例1制得的最终产物(即配合物1)和本发明实施例5 制得的最终产物(即配合物2)从交流磁化率中获得的温度依赖性弛豫时间生成的阿伦尼乌斯方程图,其中,(a)为配合物1和2在Hdc=0Oe的阿伦尼乌斯方程图,(b)为配合物1和2在Hdc=1500Oe的阿伦尼乌斯方程图。
具体实施方式
下面结合具体实施例对本发明作进一步的详述,以更好地理解本发明的内容,但本发明并不限于以下实施例。
实施例1:配合物1的制备
具体的合成方法为:将0.1mmol的2-甲基-5,7-二溴-8-羟基喹啉(0.0317g) 和0.1mmol的Dy(NO3)3·6H2O(0.0460g),加到一端封闭且长约18cm的Pyrex 管中,再加入0.1mmol的1,10-林菲罗琳(0.0182g),再加入0.4mL的1,4-二氧六环和1.1mL的水(1,4-二氧六环和水的体积比为1:2.75),然后用Et3N(约 10μL)调节体系的pH=5.5,将Pyrex管抽真空,将其另一端封口;将封好的 Pyrex管放入90℃的烘箱中保温反应72h,取出,缓慢冷却至室温,可观察到 Pyrex管底部,有黄色块状晶体析出。产率为32.60%(基于Dy)。
对本实施例所得产物进行表征:
1)单晶衍射及结构解析:
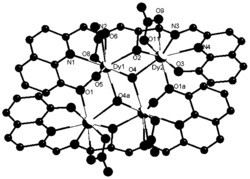
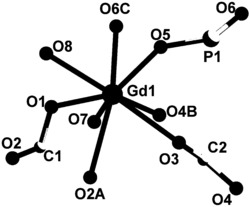
选取尺寸适中的黄色块状晶体置于安捷伦公司SuperNova单晶衍射仪上,采用石墨单色化的 射线进行单晶测试。本实施例所得产物的初始晶体结构均采用SHELXS-97直接法解出,几何加氢,非氢原子坐标及各向异性热参数采用SHELXL-97均经全矩阵最小二乘法精修。所得晶体学和结构精修数据如下述表1所示,部分键长键角数据如下述表2所示,所得浅黄色块状晶体的化学结构如图1所示。
如图1所示,配合物1的空间群是P21/c,不对称单元由一个Dy,两个 2-甲基-5,7-二溴-8-羟基喹啉,一个1,10-林菲罗琳和一个硝酸根组成。金属中心(Dy)与螯合配体提供的N1O1、N1N1和硝酸根发生配位。
表1:配合物1的晶体学数据表
表2:配合物1的部分键长键角表
2)热重分析
实验温度控制在室温到1000℃之间,流速为15cm
3)粉末衍射分析
为了研究所得产物的大量样品与单颗晶体的统一性,即纯相物质。本申请人将所得产物在常温条件下利用粉末衍射进行测试,测试范围为5-55°,扫描速率为5°/min。然后通过mercury软件模拟,将所得产物单晶结构的CIF 文件模拟得出粉末图谱,再与实际谱图相比较,可以看出特征峰的位置及峰型基本一致,表明大量物质为纯相。所得产物的粉末衍射谱图如图3所示。
通过上述表征,确定所得的黄色块状晶体为本发明所述的配合物1即 Dy
对比例1-3
重复实施例1,不同的是,反应在常温条件下进行。结果没有晶体或其它形状(如粉末)产物生成。
重复实施例1,不同的是,1,4-二氧六环和水的体积比在限定的1:2-4 范围以外。结果没有晶体或其它形状(如粉末)产物生成。
重复实施例1,不同的是,调节体系的pH值在限定的5.5-6.0范围以外。结果没有晶体或其它形状(如粉末)产物生成。
实施例2:配合物1的制备
重复实施例1,不同的是:
1)混合溶剂的组成中,1,4-二氧六环和水的体积比为1:4;
2)用三乙胺调节所得溶液的pH=5.8;
3)反应在60℃条件下进行。
反应完成后,缓慢冷却至室温,在Pyrex管底部有黄色块状晶体析出。产率为32.60%(基于Dy)。
对本实施例所得产物进行单晶衍射等分析,确定所得的黄色块状晶体为本发明所述的配合物1即Dy
实施例3:配合物1的制备
重复实施例1,不同的是:
1)混合溶剂的组成中,1,4-二氧六环和水的体积比为1:2;
2)用三乙胺调节所得溶液的pH=6.0;
3)反应在90℃条件下进行。
反应完成后,缓慢冷却至室温,在Pyrex管底部有黄色块状晶体析出。产率为32.85%(基于Dy)。
对本实施例所得产物进行单晶衍射等分析,确定所得的黄色块状晶体为本发明所述的配合物1即Dy
实施例4:配合物1的制备
1)重复实施例1,不同的是:
2)混合溶剂的组成中,1,4-二氧六环和水的体积比为1:3.5;
3)用三乙胺调节所得溶液的pH=5.6;
反应在50℃条件下进行。
反应完成后,缓慢冷却至室温,在Pyrex管底部有黄色块状晶体析出。产率为31.30%(基于Dy)。
对本实施例所得产物进行单晶衍射等分析,确定所得的黄色块状晶体为本发明所述的配合物1即Dy
实施例5:配合物2的制备
具体的合成方法为:将0.1mmol的2-甲基-5,7-二氯-8-羟基喹啉(0.0228g) 和0.1mmol的Dy(NO3)3·6H2O(0.0460g),加到一端封闭且长约18cm的Pyrex 管中,再加入0.1mmol的1,10-林菲罗琳(0.0182g),然后加入甲醇和乙腈的混合物(其中甲醇为1mL,甲醇和乙醇的体积比为1:2),然后用Et3N(约10 μL)调节体系的pH=4.1,将Pyrex管抽真空,将其另一端封口;将封好的Pyrex 管放入90℃的烘箱中保温反应72h,取出,缓慢冷却至室温,可观察到Pyrex 管底部,有黄色块状晶体析出。产率为43.47%(基于Dy)。
对本实施例所得产物进行表征:
1)单晶衍射及结构解析:
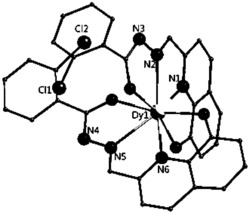
选取尺寸适中的黄色块状晶体置于安捷伦公司SuperNova单晶衍射仪上,采用石墨单色化的 射线进行单晶测试。本实施例所得产物的初始晶体结构均采用SHELXS-97直接法解出,几何加氢,非氢原子坐标及各向异性热参数采用SHELXL-97均经全矩阵最小二乘法精修。所得晶体学和结构精修数据如前述表1所示,部分键长键角数据如下述表3所示,所得浅黄色块状晶体的化学结构如图4所示。
如图4所示,配合物2的空间群是P21/c,不对称单元由一个Dy,两个 2-甲基-5,7-二氯-8-羟基喹啉,一个1,10-林菲罗琳和一个硝酸根组成。金属中心(Dy)与螯合配体提供的N1O1、N1N1和硝酸根发生配位。
表3:配合物2的部分键长键角表
2)热重分析
实验温度控制在室温到1000℃之间,流速为15cm
3)粉末衍射分析
为了研究所得产物的大量样品与单颗晶体的统一性,即纯相物质。本申请人将所得产物在常温条件下利用粉末衍射进行测试,测试范围为5-55°,扫描速率为5°/min。然后通过mercury软件模拟,将所得产物单晶结构的CIF 文件模拟得出粉末图谱,再与实际谱图相比较,可以看出特征峰的位置及峰型基本一致,表明大量物质为纯相。所得产物的粉末衍射谱图如图6所示。
通过上述表征,确定所得的黄色块状晶体为本发明所述的配合物2即 Dy
对比例4-6
重复实施例1,不同的是,反应在常温条件下进行。结果没有晶体或其它形状(如粉末)产物生成。
重复实施例1,不同的是,甲醇和乙腈的体积比在限定的1:1-3范围以外。结果没有晶体或其它形状(如粉末)产物生成。
重复实施例1,不同的是,调节体系的pH值在限定的4.0-4.5范围以外。结果没有晶体或其它形状(如粉末)产物生成。
实施例6:配合物2的制备
重复实施例5,不同的是:
1)混合溶剂的组成中,甲醇和乙腈的体积比为1:3;
2)用三乙胺调节所得溶液的pH=4.2;
3)反应在120℃条件下进行。
反应完成后,缓慢冷却至室温,在Pyrex管底部有黄色块状晶体析出。产率为43.21%(基于Dy)。
对本实施例所得产物进行单晶衍射等分析,确定所得的黄色块状晶体为本发明所述的配合物2即Dy
实施例7:配合物2的制备
重复实施例5,不同的是:
1)混合溶剂的组成中,甲醇和乙腈的体积比为1:1;
2)用三乙胺调节所得溶液的pH=4.5;
3)反应在50℃条件下进行。
反应完成后,缓慢冷却至室温,在Pyrex管底部有黄色块状晶体析出。产率为43.55%(基于Dy)。
对本实施例所得产物进行单晶衍射等分析,确定所得的黄色块状晶体为本发明所述的配合物2即Dy
实施例8:配合物2的制备
重复实施例5,不同的是:
1)混合溶剂的组成中,甲醇和乙腈的体积比为1:3;
2)用三乙胺调节所得溶液的pH=4.0;
3)反应在90℃条件下进行。
反应完成后,缓慢冷却至室温,在Pyrex管底部有黄色块状晶体析出。产率为42.85%(具有Dy)。
对本实施例所得产物进行单晶衍射等分析,确定所得的黄色块状晶体为本发明所述的配合物2即Dy
对本发明所述的配合物1和2(分别按实施例1和实施例5所述方法制得) 进行荧光光谱分析:
配合物1和2,有两个不同的激发峰,波长分别为420nm和330nm,归因于有机配体从基态电平(π1)到激发态电平(π1
对本发明所述的配合物1和2(分别按实施例1和实施例5所述方法制得) 进行磁性分析
为了探索各向异性磁矩的磁化动力学,对两个配合物进行交流(AC)磁化率测试,在0直流场和一个3Oe交流场中以1-1000Hz范围内的频率振荡。对于配合物1和2,χ”与T的关系曲线在不同频率下从20到2K测量;实部(χ') 和虚部(χ”)部分都具有频率和温度依赖性,分别如图9和10所示。配合物 1和配合物2在所有测试频率处的χ'值在冷却至2K时连续增加,没有任何最大值,表明其能垒很小,预期阻塞温度位于2K以下。此外,除了1Hz以外,在所有应用的频率中出现配合物1和2的异相峰,表明预期会有更高的能垒。此外,配合物1和2的频率相关AC数据在不同温度下不存在DC场的情况下表征;χ”曲线的峰值以频率序列从低到高逐渐转移,说明配合物2的χ”在选定的温度范围内总是表现出频率依赖性。弛豫时间τ值取决于频率相关的交流磁化率(如图11所示),由阿伦尼乌斯拟合后的χ'最大值[τ=τ0 exp(Ueff/kBT)]得出,能垒和弛豫时间分别为Ueff=0.60K和τ0=1.23×10
为了抑制量子隧道效应,两个配合物的AC磁数据测量在额外的1500Oe DC场下进一步进行。在相同频率1,10,100,300,499,700和999Hz下测试1 和2的交流磁化率曲线的依赖性(如图12所示)。两个配合物的χ'和χ”磁化率在较低温度区域显示出显着的热依赖性最大值,明确表明缓慢的磁松弛,并且通过QTM过程的可能松弛行为在1500Oe。同时,对频率相关的交流磁数据实验进行了表征,对于配合物2所有频率的依赖性出现在6-17K,配合物1所有频率的依赖性出现在6-15K。显然,随着温度的升高,两种配合物中χ'曲线的最大值从较低频率缓慢移动到较高频率(如图13所示)。此外,ln (τ)形式的磁化弛豫时间(τ)被描述为图13中1/T的函数。根据高温曲线拟合Arrhenius定律的行为,可以获得有效屏障(Ueff/kB)。在1500Oe直流场下,Ueff=92.8K,τ0=3.8×10
8-羟基喹啉衍生物和1,10-邻菲啰啉混合掺杂的镝配合物及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。










动态评分
0.0