










专利摘要
本发明提供了抑制了凝胶化,包含高分支型超高分子量共聚物及线状聚合物的苯乙烯系树脂组合物及其制造方法。其为制造含有高分支型超高分子量共聚物和线状聚合物的苯乙烯系树脂组合物的方法,以苯乙烯为必须的单乙烯基化合物中,添加以重量基准计在平均1个分子中具有乙烯基2个以上、具有分支结构的溶剂可溶性多官能乙烯基共聚物50ppm~5000ppm,进行聚合反应,获得包含该溶剂可溶性多官能乙烯基共聚物与该单乙烯基化合物发生聚合生成的高分支型超高分子量共聚物、该单乙烯基化合物发生聚合生成的线状聚合物的苯乙烯系树脂组合物。
权利要求
1.苯乙烯系树脂组合物的制造方法,其为制造含有高分支型超高分子量共聚物和线状聚合物的苯乙烯系树脂组合物的方法,其特征在于,向以苯乙烯为必须的单乙烯基化合物添加以重量基准计在平均1分子中具有乙烯基2个以上、具有分支结构的溶剂可溶性多官能乙烯基共聚物50ppm~5000ppm,进行聚合反应,获得包含该溶剂可溶性多官能乙烯基共聚物和该单乙烯基化合物共聚生成的高分支型超高分子量共聚物、该单乙烯基化合物聚合生成的线状聚合物的苯乙烯系树脂组合物。
2.权利要求1所述的苯乙烯系树脂组合物的制造方法,其特征在于,溶剂可溶性多官能乙烯基共聚物为聚合可与二乙烯基化合物共聚的单乙烯基化合物而获得,进一步在结构单元中作为摩尔分数以0.05~0.50的范围含有由下述式(a1)表示的含有二乙烯基化合物来源的侧链乙烯基的单元,在重均分子量中的惯性半径(nm)和上述摩尔分数的比为1~100的范围内,
(式中,R1表示来源于二乙烯基化合物的烃基)。
3.苯乙烯系树脂组合物,其为根据权利要求1或2所述的方法获得的苯乙烯系树脂组合物,其特征在于,含有重均分子量为1,000,000以上的高分支型超高分子量共聚物2.0~20.0重量%和重均分子量为100,000~500,000的线状苯乙烯系聚合物80.0~98.0重量%,重均分子量为200,000~800,000。
说明书
技术领域
本发明涉及苯乙烯系树脂组合物的制造方法及通过该制造方法获得的苯乙烯系树脂组合物,所述苯乙烯系树脂组合物包含如下的混合物:添加混合含有苯乙烯的乙烯性不饱和单体和在一分子内具有多个双键的溶剂可溶性多官能乙烯基共聚物,使之进行聚合而获得的高分支型超高分子量成分和线状成分的混合物。
背景技术
苯乙烯系树脂由于廉价,且透明性、耐热性、机械强度等方面优异,而且成形性良好,因此在电气制品、家庭用品等多个领域中广泛使用。这些成形品通过注塑成形、或由片材经真空、压力成形,进一步由挤压机,将树脂挤出为被称为型坯的筒状,挟入模具中,之后从内部吹入压缩空气等的吹塑成形等手段而获得。另外,为了获得轻量且具有隔热性能的成形体,也使用了发泡成形等技术。苯乙烯树脂的发泡体具有轻量性、隔热性、缓冲性等多个特征,从以住宅用隔热材料为代表的聚苯乙烯泡沫到热成形为盘、碗等,用作食品包装的片材状聚苯乙烯纸,或使通过悬浊聚合而获得的粒子状态的树脂直接含浸戊烷等脂族烃,通过蒸汽等的加热形成容器的珠发泡等技术广泛使用。在这些成形方法中,特别是具有熔融压延过程的片材成形、吹塑成形、发泡成形等成形方法对熔融时的应变固化性高的原料的要求高。
在上述成形方法中,作为使用应变固化性低的树脂材料时的问题,可举出,在片材成形中,对食品容器等深加工制品进行二次加工时,因伴随加热熔融的下垂现象,制品容易产生厚度不均,另外由于压延性不足导致的制品的裂纹、破损等变得容易发生;吹塑成形中,在型坯成形时,如果应变固化性低,则不仅产生缩坯,成形变得困难,而且由于厚度不均导致的制品强度的不均匀大;进一步因为发泡成形中提高绝热性能,所以使发泡体的气泡微小化、独立化变得困难等现象。在发泡成形中,为了提高独立气泡的比率,为了使压延时在壁面不存在极薄的部分,适用可压延的材料,因为应变固化性小的材料,对薄壁化区域的压延的阻力小,所以如果一旦产生薄的部分,则陷入进一步被压延更加薄壁化的恶性循环中,最终以至于壁面的断裂。应变固化性大的材料,被压延的区域的粘度上升,对薄壁化的部分的压延的阻力也比厚壁部变高,所以不会陷入上述恶性循环中,以均匀的膜厚的压延成为可能。
作为提高熔融状态下的张力、应变固化性等熔融特性的手段,以往已知使苯乙烯系树脂组合物含有超高分子量成分的方法是有效的。
作为获得含有超高分子量成分的树脂组合物的方法,已知在一定范围内含有专利文献1中记载的分子量为200万以上的成分的苯乙烯系聚合物组合物。但是,作为获得该组合物的方法,虽然提出了在低温下使用本体聚合、溶液聚合的手段进行聚合,生成超高分子量成分,或在熔融状态下混合通过阴离子聚合、乳液聚合等其他途径进行了调整的超高分子量成分的方法,但该方法存在生产性差、掺混其他途径聚合的成分的情况下成本变高等问题。
为了避免上述问题,提出了例如在一定范围内含有记载于专利文献2中的含有多官能乙烯基化合物单元100万以上的分子量成分的苯乙烯系聚合物等,为了含有分支型超高分子量成分,向乙烯系单体添加极少量以芳香族二乙烯基化合物为代表的芳香族多官能乙烯基化合物进行聚合。但是,如果将该手段应用于连续本体聚合,继续长期的反应的情况下,在存在于聚合反应器的壁面的被称为境膜的流动停止的区域中,存在凝胶化进行这样的问题,为了避免上述问题,则多官能芳香族乙烯基化合物的添加量受到限制,难以生成所希望的量的超高分子量成分。还有,悬浊聚合时,因为完成聚合至几乎没有未反应的单体这样的特征,所以如果原封不变地适用所提出的多官能乙烯化合物,则聚合结束时,组入聚合物链中的多官能乙烯基化合物来源的侧链乙烯基在转化率90%以上的区域急速反应,显著高分子量化,所以分子量、分子量分布的控制困难。
进一步,虽然专利文献3中公开了利用悬浊聚合,使用多官能聚合引发剂,使苯乙烯系聚合物含有具有分支结构的超高分子量成分的方法,在专利文献4中也公开了使用多官能聚合引发剂,使苯乙烯聚合物含有具有分支结构的超高分子量成分的方法,但在该方法中,苯乙烯系聚合物整体容易高分子量化,如果为了避免该情况,同时使用链转移剂等分子量调节剂,效果容易变得不充分。另外,专利文献5中记载了通过同时使用多官能芳香族乙烯基化合物和链转移剂,控制苯乙烯系树脂的聚合度的方法,但不仅存在与使用多官能引发剂的情况同样的抵消效果的问题,而且存在作为链转移剂,如果使用硫醇类,则由于特有的臭气的问题,存在使用范围受到限制这样的问题。
现有技术文献
专利文献
专利文献1:特公昭62-61231号公报
专利文献2:特开平2-170806号公报
专利文献3:特开平7-278218号公报
专利文献4:特开平8-59721号公报
专利文献5:特开2002-241413号公报
发明内容
本发明的目的是提供效率良好地制造苯乙烯系树脂组合物的方法以及利用该方法获得的熔融特性优异的含有高分支型超高分子量材料的苯乙烯系树脂组合物,其中该苯乙烯系树脂组合物具备对在片材成形、发泡成形、吹塑成形等成形加工时需要熔融压延过程的加工方法最适合的熔融特性、无凝胶状物,含有高分支型超高分子量成分和线状成分。
即,本发明涉及苯乙烯系树脂聚合物的制造方法,为制造含有高分支型超高分子量共聚合物和线状聚合物的苯乙烯系树脂组合物的方法,该苯乙烯树脂组合物包含通过向以苯乙烯为必须的单乙烯基化合物中添加混合以重量基准计在平均1分子中具有乙烯基2个以上、具有分支结构的溶剂可溶性多官能乙烯基共聚物50ppm~5000ppm而进行聚合,该溶剂可溶性多官能乙烯基共聚物和该单乙烯基化合物发生聚合生成的高分支型超高分子量共聚物和该乙烯系单体聚合产生的线状聚合物。
在上述制造方法中,作为溶剂可溶性多官能乙烯基共聚物,优选举出使二乙烯基化合物和可共聚的单乙烯基化合物聚合而获得,而且在结构单元中作为摩尔分数计以0.05~0.50的范围含有以下式(a1)表示的二乙烯基化合物来源的侧链乙烯基,在重均分子量中的惯性半径(nm)与上述摩尔分数的比为1~100的范围内的多官能乙烯基共聚物。
(式中,R1表示来源于二乙烯基化合物的烃基。)
另外,本发明涉及含有高分支型超高分子量共聚物的苯乙烯系树脂组合物,其特征在于,其含有利用上述制造方法而获得的重均分子量为1,000,000以上的高分支型超高分子量共聚物2.0~20.0重量%、重均分子量为100,000~500,000的线状苯乙烯系聚合物80.0~98.0重量%,重均分子量为200,000~800,000。
具体实施方案
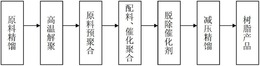
以下,详细地说明本发明。作为本发明中使用的聚合方法,优选使用所谓连续本体聚合法:添加混合包含苯乙烯的单乙烯基化合物和溶剂可溶性多官能乙烯基共聚物,根据需要的溶剂、聚合催化剂、链转移剂等,连续地将单体类送入具备串联和/或并联排列的1个以上的反应器和除去未反应单体等的挥发成分去除工序的设备,阶段性地进行使之聚合。作为反应器的样式,例示了完全混合型的槽型反应器、具有活塞流性的塔型反应器、一边进行聚合一边排出一部分聚合液的循环型反应器等。这些反应器的排列顺序不受特别限制,但在连续生产中为了抑制凝胶状物的生成,在溶剂可溶性多官能乙烯基共聚物为未反应的状态下,在反应器壁面的境膜中不出现高浓度地滞留的状态是重要的,作为第一反应器,优选地选择完全混合型的槽型反应器。
另外,作为本发明的聚合方法,也优选使用所谓悬浊聚合法:添加混合包含苯乙烯的单乙烯基化合物、溶剂可溶性多官能乙烯基共聚物、根据需要的聚合催化剂、链转移剂等之后,使其悬浊于水中,进行聚合。为了使分散稳定化,向溶解有聚乙烯醇、甲基纤维素等有机系分散剂,或磷酸三钙、硫酸镁等无机系分散剂、十二烷基苯磺酸钠等阴离子性表面活性剂的水中投入单体类,在搅拌下使之分散,在100~150℃的范围内进行聚合。如果考虑反应结束后直接在水分散状态下不变,使之在加压下含浸戊烷等脂肪族烃发泡气体的情况,则优选反应结束时的最终的聚合转化率为99%以上。如果不足99%,则在二次成形时的温度为残存单体的沸点以上的情况下,伴随显著的恶臭。
在本发明中,为了使最终的聚合转化率为99%以上,优选对相对于原料装入量,添加了1小时半衰期温度为130±10℃的范围的过氧化物系催化剂200ppm以上而成的原料溶液在反应温度120℃以下进行聚合至50%以上之后,以超过上述催化剂的半衰期温度5℃以上的反应温度,进行3小时以上的聚合。通过使用该条件,不会极端地延长聚合时间,而可容易地使最终的聚合转化率为99%以上。在后段的反应温度为1小时半衰期温度以下的情况下,聚合时间极端地延长,使生产性显著降低。
为了使最终转化率为99%以上,作为所使用的有机过氧化物引发剂,例示了叔丁基过氧化醋酸酯、叔丁基过氧化苯甲酸酯、2,2-二-(叔丁基过氧)丁烷、过氧化二枯基等。
在本发明中,溶剂可溶性多官能乙烯基共聚物可在溶解于单乙烯基化合物类、聚合溶剂等的状态下,根据需要,在连续本体聚合情况下多个反应器的中途,在悬浊聚合时聚合反应的中途添加。
在本发明中使用的以苯乙烯为必须的单乙烯基化合物(以下也称之为苯乙烯系单体)可以是苯乙烯为100%,也可以是包含苯乙烯和其他的单乙烯基化合物的混合物。作为其他的单乙烯基化合物,可举出具有可与苯乙烯共聚的烯属双键的物质:p-甲基苯乙烯等芳香族乙烯系单体类;丙烯酸、甲基丙烯酸等丙烯酸单体;丙烯腈、甲基丙烯腈等乙烯基氰单体;丙烯酸丁酯、甲基丙烯酸甲酯等丙烯酸系单体或无水马来酸、富马酸等α,β-乙烯不饱和羧酸类;苯基马来酰亚胺、环己基马来酰亚胺等酰亚胺系单体类。可同时使用这些以外的单乙烯基化合物1种或2种以上。因此,由于苯乙烯和其他的单乙烯基化合物的比例为苯乙烯20~100摩尔%、其他的单乙烯基化合物为0~80摩尔%发挥了苯乙烯系树脂组合物的特性,所以优选。
本发明中使用的溶剂可溶性多官能乙烯基共聚物(以下也称之为多官能乙烯基共聚物)为通过和苯乙烯系单体进行共聚,分支地产生被分支的超高分子量的苯乙烯系树脂的物质。
本发明中使用的多官能乙烯基共聚物可根据特开2004-123873号公报、特开2005-213443号公报、WO2009/110453等中公开的方法获得。具体地讲,使用二乙烯基化合物和至少一种以上的单乙烯基化合物,使之共聚,获得具有以式(a1)表示的反应性的侧链乙烯基的共聚物。进一步,如上述专利文献所记载的,可使用末端上被导入乙烯基以外的其他末端基的物质,特别是因像苯氧基丙烯酸甲酯类这样的分子内具有不饱和键的化合物而被末端改性的物质除了(a1)以外还可作为交联点而作用,所以优选。此时,因为含有末端的不饱和键的结构单元(a2)也具有乙烯基,所以与式(a1)的结构单元的合计的摩尔分数(a3)显示了整体的乙烯基的存在量。
为了获得多官能乙烯基共聚物,作为所使用的二乙烯基化合物,例示了以二乙烯基苯为代表的二乙烯基芳香族化合物类和以乙二醇二(甲基)丙烯酸酯为代表的脂肪族、脂环式(甲基)丙烯酸酯类等。
另外,作为在此使用的单乙烯基化合物,可举出如前述的包含苯乙烯等的单乙烯基芳香族化合物的单乙烯基化合物。
作为多官能乙烯基共聚物的制造方法,可通过例如在选自路易斯酸催化剂、选自酯化合物的助催化剂的存在下,使选自二乙烯基芳香族化合物、单乙烯基芳香族化合物及其他单乙烯基化合物的两种以上的化合物进行阳离子共聚而获得。另外,使用(甲基)丙烯酸酯系的二乙烯基、单乙烯基化合物的情况下,因为阳离子反应不进行,所以可在过氧化物等自由基催化剂的存在下,通过自由基聚合而获得。
二乙烯基化合物和单乙烯基化合物的使用量,以产生在本发明中使用的多官能乙烯基共聚物的组成的方式而决定,但优选使用全部单体的10~90摩尔%、更优选30~90摩尔%、更加优选50~90摩尔%的二乙烯基化合物。优选使用全部单体的90~10摩尔%、更优选70~10摩尔%、更加优选50~10摩尔%的单乙烯基化合物。在此,像2-苯氧基乙基丙烯酸甲酯这样的在阳离子聚合中,作为末端改性剂而起作用的物质不作为单体进行计算。
作为在多官能乙烯基共聚物的制造中使用的路易斯酸催化剂,为包含金属离子(酸)和配位体(碱)的化合物,如果是可接受电子对的物质则可不受特别限制地使用。从分子量和分子量分布的控制及聚合活性的观点来说,最优选使用三氟化硼的醚(二乙醚、二甲醚等)络合物。路易斯酸催化剂相对于全部单体1摩尔,在0.001~10摩尔的范围内使用,但更优选0.001~0.01摩尔。如果路易斯酸催化剂的使用量过大,则因为聚合速度变得过快,所以分子量分布的控制变得困难,因此不优选。
作为助催化剂,可举出选自酯化合物的1种以上。其中从聚合速度及共聚物的分子量分布控制的观点来说,优选使用碳数4~30的酯化合物。从容易获得的观点来说,优选使用乙酸乙酯、乙酸丙酯及乙酸丁酯。助催化剂相对于单体化合物1摩尔,在0.001~10摩尔范围内使用,但更优选0.01~1摩尔。如果助催化剂的使用量过大,则聚合速度降低,共聚物的产率下降。另一方面,如果助催化剂的使用量过小,则聚合反应的选择性降低,除了产生分子量分布的增大,凝胶的生成等之外,聚合反应的控制也变得困难。
另外,作为在通过自由基聚合制造多官能乙烯基共聚物时使用的催化剂,例示了像以偶氮二异丁腈为代表的偶氮化合物、过氧化二苯甲酰、叔丁基过氧苯甲酸酯等单官能性的过氧化物或1,1-双(叔丁基过氧化)环己烷这样的2官能性以上的多官能性的过氧化物,可将其单独使用或同时使用两种以上。
在本发明中使用的多官能乙烯基共聚物可通过如上述的制造方法而获得,但需要作为单体而使用的二乙烯基化合物的乙烯基的一部分不聚合而残留。因此,平均在1分子中存在至少2个以上,优选3个以上的乙烯基。该乙烯基主要作为以上式(a1)表示的结构单元而存在。因此,通过乙烯基的一部分不发生聚合而残留,可抑制交联反应,赋予溶剂可溶性。在此,所谓溶剂可溶性是指可溶于甲苯、二甲苯、四氢呋喃(THF)、二氯乙烷或氯仿,具体地是说在25℃时在这些溶剂100g中溶解5g以上,不产生凝胶。另一方面,二乙烯基化合物的一部分由于两个乙烯基反应而交联或分支是必要的,由此可成为具有分支结构的共聚物。如此,对于二乙烯基化合物的一部分,使2个乙烯基的一个发生反应,一个不聚合而残留,对于其他的一部分,使2个乙烯基发生反应,由此可获得在本发明中使用的多官能乙烯基共聚物。像这样的获得多官能乙烯基共聚物的聚合方法,如上所述为公知的,能够以使如上所述的方式而制造。
多官能乙烯基共聚物的重均分子量(Mw)优选为1,000~100,000,更优选5,000~70,000。小于1000的情况,与使用芳香族二乙烯基化合物或多官能(甲基)丙烯酸酯类的情况同样,在连续聚合中抑制凝胶化进行的效果减小,在悬浊聚合中在高转化率区域中的分子量分布的控制也变得困难,不能获得充分的效果,因此不优选。
被导入多官能乙烯基共聚物的含有二乙烯基化合物来源的乙烯基的单元具有以上述式(a1)表示的结构单元,但该结构单元(a1)的摩尔分数为0.05~0.50。少于0.05摩尔的情况下,难以获得高分支型超高分子量共聚物,因此不优选。另一方面,超过0.50摩尔的情况下,高分支型超高分子量共聚物的分子量过度增大,凝胶化变得容易发生,因此不优选。另外,如上所述,因为通过分子内具有不饱和键的化合物而末端改性的物质除了以式(a1)表示的结构单元,含有末端的不饱和键的结构单元(a2)也具有乙烯基,所以两者合计的摩尔分数(a3)可以为0.05~0.50。
另外,多官能乙烯基共聚物优选其重均分子量中的惯性半径(nm)与上述结构单元(a1)的摩尔分数或上述合计的摩尔分数(a3)的比为1~100的范围。为了不伴随将用于赋予应变固化性的分支型超高分子量成分凝胶化而进行调制,更优选10~80的范围。上述的比超过100的情况下,凝胶化不进行,但难以获得高分支型超高分子量共聚物,因此不优选。另一方面,小于1的情况下,高分支型超高分子量共聚物的分子量过度增大,容易发生凝胶化,因此不优选。在此,惯性半径为利用实施例中记载的方法而测定的值。另外,多官能乙烯基共聚物为具有分子量方面的分布的聚合物,当然,因为该惯性半径也具有分布,所以采用重均分子量中的惯性半径作为整体的惯性半径的平均值。
在此所定义的表示惯性半径与双键的含量的指标,即结构单位(a1)的摩尔分数或上述合计的摩尔分数(a3)的比可以说为在构成分支型超高分子量成分时,表示成为核的多官能乙烯基共聚物在聚合反应溶液中多大范围中,具有什么程度的反应点的指标。如果该比过小的话,则反应点位于附近,变得容易引起凝胶化,另外,如果该比过大,则分支型成分的高分子量化变得困难。
作为多官能乙烯基共聚物相对于苯乙烯系单体的配合率,以重量基准计,优选50ppm~5000ppm,更优选100ppm~3000ppm。多官能乙烯基共聚物的配合率小于50ppm的情况下,难以获得本发明的充分的效果,因此不优选。另一方面,超过5000ppm的情况下,存在产生凝胶的可能性。
通过使前述多官能乙烯化合物共聚物和苯乙烯系单体聚合,获得了作为多官能乙烯基共聚物和苯乙烯系单体的共聚物的高分支型超高分子量共聚物(也称为高分支型共聚物)和仅由苯乙烯系单体生成的线状聚合物的混合物,即本发明的苯乙烯系树脂组合物。作为苯乙烯系单体,使用2种以上的单体的情况下,线状聚合物变为共聚物。
通过本发明获得的苯乙烯系树脂组合物的重均分子量(Mw)优选为20万~80万。Mw不足20万的话,加工之后的冲击强度不充分,Mw超过80万的话,粘度上升,加工性变得不充分。
在如上所述的苯乙烯系树脂组合物中,包含高分支共聚物和线状聚合物,但通过使之为显示如上所述的Mw的苯乙烯系树脂组合物,高分支共聚物的Mw变为100万以上的超高分子量,线状聚合物变为10万~50万。因此,优选Mw为100万以上的高分支超高分子量共聚物与Mw为10万~50万的线状苯乙烯系聚合物的比例为2:98~20:80。这些的比例,可通过调整多官能乙烯基共聚物相对于苯乙烯系单体的配合比例和聚合条件,进行控制。
关于苯乙烯系树脂组合物的制造,从聚合反应的控制的观点出发,根据需要,可使用聚合溶剂、有机过氧化物等聚合引发剂或脂肪族硫醇等链转移剂。
聚合溶剂在连续本体聚合中,为了降低反应物的粘性而使用,作为该有机溶剂,可举出甲苯、乙苯、二甲苯、乙腈、苯、氯苯、二氯苯、苯甲醚、氰基苯、二甲基甲酰胺、N,N-二甲基乙酰胺、甲基乙基酮等。
特别是在连续本体聚合中,在想要使多官能乙烯基共聚物的添加量变多的情况下,从抑制凝胶化的观点来说,也优选使用有机溶剂。由此,可使先前所示的多官能乙烯基共聚物的添加量飞跃性地增加,难以产生凝胶。
有机溶剂的使用量不受特别限定,但从控制凝胶化这样的观点出发,通常优选相对于单体成分的合计量100重量份,为1~50重量份,更优选5~30重量份的范围内。超过50重量份的情况下,生产性显著降低,链状苯乙烯系树脂的分子量过度降低,因此不优选。
作为聚合引发剂,优选自由基聚合引发剂,可举出公知常用的例如1,1-二(叔丁基过氧基)环己烷、2,2-二(叔丁基过氧基)丁烷、2,2-二(4,4-二-丁基过氧基环己基)丙烷等过氧化缩酮类;异丙苯氢过氧化物、叔丁基氢过氧化物等氢过氧化物类;过氧化二叔丁基、过氧化二枯基、二-叔己基过氧化物等二烷基过氧化物类;过氧化苯甲酰、二酰基过氧化物等二酰基过氧化物类;叔丁基过氧化苯甲酸酯、二-叔丁基过氧间苯二甲酸酯、过氧叔丁基丙基单碳酸酯等过氧化酯类;N,N'-偶氮二异丁腈、N,N'-偶氮双(环己烷-1-甲腈)、N,N'-偶氮双(2-甲基丁腈)、N,N'-偶氮双(2,4-二甲基戊腈)、N,N'-偶氮双[2-(羟甲基)丙腈]等,可使用这些的1种或组合使用2种以上。
以使苯乙烯系树脂组合物的分子量不会变得过大的方式而添加链转移剂,可使用具有1个链转移基的单官能链转移剂、具有多个链转移基的多官能链转移剂。作为单官能链转移剂,可举出烷基硫醇类、巯基乙酸酯类等。
作为多官能链转移剂,可举出通过硫代乙醇酸或3-巯基丙酸而酯化乙二醇、新戊二醇、三羟甲基丙烷、季戊四醇、二季戊四醇、三季戊四醇、山梨糖醇等多元醇羟基的物质。
实施例
以下使用实施例进一步具体地说明本发明。所使用的测定方法如下所述。
(GPC测定法)通过高速液相色谱仪(Tosoh Corporation制HLC-8220GPC)、RI检测器、TSK凝胶GMH×1×2、溶剂四氢呋喃、流速1.0ml/分钟、40℃温度时测定标准聚苯乙烯换算的平均分子量。
(双键定量法)结构单元(a1)、末端改性剂来源的双键(a2)及两者的合计的摩尔分数(a3)为使用日本电子制JNM-LA600型核磁共振分光装置,通过13C-NMR及1H-NMR分析,确定结构。作为溶剂,使用氯仿-d1,将四甲基硅烷的共振线作为内部标准而使用。
(惯性半径)将样品调整为0.5%的四氢呋喃溶液之后,通过膜过滤器进行过滤,使用GPC多角度光散射法对滤液进行测定。进一步,将样品调整为0.2%四氢呋喃溶液之后放置1天。其后,使用四氢呋喃稀释为4种浓度(0.02、0.05、0.10、0.12重量%)的溶液,使用这些溶液,进行dn/dc测定,由获得的dn/dc值算出样品的惯性半径。
(凝胶状物的确认)使用注塑成形机,成形为180mm×180mm×3mm的平板,通过目视,确认在含有凝胶状物时产生的来自门部分的线状痕迹的有无。
合成例1
(多官能乙烯基共聚物α)
将二乙烯苯3.1摩尔(399.4g)、乙基乙烯苯0.7摩尔(95.1g)、苯乙烯0.3摩尔(31.6g)、2-苯氧基乙基甲基丙烯酸酯2.3摩尔(463.5g)、甲苯974.3g投入3.0L的反应器内,在50℃时添加42.6g的三氟化硼二乙醚络合物,使之反应6.5小时。通过碳酸氢钠溶液使聚合反应停止之后,通过纯水洗涤油层3次,在室温时将反应混合液投入大量的甲醇中,使聚合物析出。通过甲醇将所获得的聚合物洗涤、过滤、干燥、称量,获得多官能乙烯基共聚物α372.5g。该多官能乙烯基共聚物α的重均分子量Mw为8000,含有二乙烯基化合物来源的乙烯基的结构单位(a1)的摩尔分数为0.44、末端的2-苯氧基乙基甲基丙烯酸酯来源的双键(a2)为0.03、组合两者的合计的摩尔分数(a3)为0.47。另外,重均分子量8000中的共聚物的惯性半径(r)为6.4nm。与直链型的分子量8000的惯性半径为15nm相比,可知本合成例中的多官能乙烯基共聚物具有分支结构。另外,计算惯性半径(r)与摩尔分数(a3)的比(r/a3)为13.6。
合成例2
(多官能乙烯基共聚物β)
将二乙烯苯2.6摩尔(332.0g)、乙基乙烯苯1.5摩尔(198.0g)、苯乙烯1.1摩尔(109.6g)、2-苯氧基乙基甲基丙烯酸酯3.1摩尔(630.4g)、甲苯886.0g投入3.0L的反应器内,在50℃时添加35.5g的三氟化硼乙醚络合物,使之反应5.0小时。通过碳酸氢钠溶液使聚合反应停止之后,通过纯水洗涤油层3次,在室温时将反应混合液投入大量的甲醇中,使聚合物析出。通过甲醇将所获得的聚合物洗涤、过滤、干燥、称量,获得多官能乙烯基共聚物β564.0g。该多官能乙烯基共聚物β的Mw为5000,含有二乙烯基化合物来源的乙烯基的结构单元(a1)的摩尔分数为0.25,末端的2-苯氧基乙基甲基丙烯酸酯来源的双键(a2)为0.02,组合两者的合计的摩尔分数(a3)为0.27。另外,重均分子量中的共聚物的惯性半径为8.1nm。与直链型的分子量5000中的惯性半径为12nm相比,可知本合成例中的多官能乙烯基共聚物具有分支结构。
合成例3
(多官能乙烯基共聚物γ)
将二乙烯苯1.2摩尔(159.8g)、乙基乙烯苯0.7摩尔(95.3g)、苯乙烯2.1摩尔(223.2g)、2-苯氧基乙基甲基丙烯酸酯3.1摩尔(632.0g)、甲苯1082.5g投入3.0L的反应器内,于50℃时添加56.8g的三氟化硼二乙醚络合物,使之反应6.0小时。通过碳酸氢钠溶液使聚合反应停止之后,通过纯水洗涤油层3次,在室温时将反应混合液投入大量的甲醇中,使聚合物析出。通过甲醇将获得的聚合物洗涤、过滤、干燥、称量,获得多官能乙烯基共聚物γ340.8g。该多官能乙烯基共聚物γ的Mw为5000,含有二乙烯芳香族化合物来源的乙烯基的结构单元(a1)的摩尔分数为0.13、末端的2-苯氧基乙基甲基丙烯酸酯来源的双键(a2)为0.01、组合两者合计的摩尔分数(a3)为0.14。另外,重均分子量中的共聚物的惯性半径为10.6nm。与直链型的分子量5000中的惯性半径为12nm相比,可知本合成例中的多官能乙烯基共聚物具有分支结构。
上述合成例1~3中的多官能乙烯基共聚物均可溶于甲苯、二甲苯、四氢呋喃、二氯乙烷及氯仿。
实施例1
均匀地混合苯乙烯85重量份、乙苯15重量份、多官能乙烯基共聚物(α)0.06重量份之后,以15L/小时连续地送入连续本体聚合设备,所述本体聚合设备具有2个串联地连接的槽型反应器、内藏有静态混合机的内容积15L的塔型反应器、以及具有预热器和真空槽的闪蒸室型的挥发成分除去装置,槽型反应器具有内容积30L的完全混合性,静态混合机具有活塞流性。以使第一反应器为130℃、第二反应器为140℃、第三反应器的入口部为140℃、出口部为160℃的方式阶段性地提高温度之后,移送至加热至220℃的预热器,投入在将压力调整为8托的预热器的正下方的真空槽,由此除去未反应单体、溶剂,之后一边从真空槽利用齿轮泵,将树脂拉拔为缕状,一边进行剪切,由此获得苯乙烯系树脂组合物。到达保持该稳定状态不变的稳定状态之后,对24小时、72小时、144小时之后的树脂组合物实施分子量、凝胶状物的评价,结果显示于表1中。
实施例2
除了代替实施例1中的多官能乙烯基共聚物(α),使用多官能乙烯基共聚物(β)之外,与实施例1同样地操作,获得聚苯乙烯树脂聚合物。表1中显示了各个时间的分子量、凝胶状物的评价结果。
实施例3
除了代替实施例1中的多官能乙烯基共聚物(α),使用多官能乙烯基共聚物(γ)之外,与实施例1同样地操作,获得聚苯乙烯树脂聚合物。表1中显示了各个时间的分子量、凝胶状物的评价结果。
实施例4
除了使实施例1中的多官能乙烯基共聚物(α)的添加量0.06重量份变为0.01重量份之外,与实施例1同样地操作,获得聚苯乙烯树脂组合物。表1中显示了各个时间的分子量、凝胶状物的评价结果。
实施例5
除了使苯乙烯为70重量份、乙苯为30重量份,实施例1中的多官能乙烯基共聚物(α)的添加量0.06重量份变为0.3重量份之外,与实施例1同样地操作,获得聚苯乙烯树脂聚合物。表1中显示了各个时间的分子量、凝胶状物的评价结果。
实施例6
除了使实施例1中的多官能乙烯基共聚物(α)的添加量0.06重量份变为0.1重量份,与苯乙烯同时,添加叔十二烷基硫醇(tDM)0.05重量份之外,与实施例1同样地操作,获得聚苯乙烯树脂组合物。
比较例1
除了不添加多官能乙烯基共聚物(α)以外,与实施例1同样地操作,获得线状聚苯乙烯。
比较例2
除了使实施例1中的多官能乙烯基共聚物(α)的添加量0.06重量份变为0.001重量份以外,与实施例1同样地操作,获得聚苯乙烯树脂组合物。
比较例3
除了使实施例1中的多官能乙烯基共聚物(α)的添加量0.06重量份变为1重量份以外,与实施例1同样地操作,获得聚苯乙烯树脂组合物。
比较例4
除了代替实施例1中的多官能乙烯基共聚物(α),使二乙烯苯为0.05重量份以外,与实施例1同样地操作,获得聚苯乙烯树脂组合物。表1中显示了各个时间的分子量、凝胶状物的评价结果。24小时未观测到凝胶状物,但72小时产生了凝胶状物,144小时变为大量含有凝胶状物的状态。
比较例5
除了代替实施例1中的多官能乙烯基共聚物(α),使二乙烯苯为0.025重量份以外,与实施例1同样地操作,获得聚苯乙烯树脂组合物。72小时未观测到凝胶状物,但确认144小时产生凝胶状物。
比较例6
除了代替实施例1中的多官能乙烯基共聚物(α),使用二乙烯苯0.05重量份,使苯乙烯与溶剂乙苯的比率为苯乙烯70重量份、乙苯30重量份以外,与实施例1同样地操作,获得聚苯乙烯树脂组合物。
将实施例1~6及比较例1~6中的反应原料的使用量及聚苯乙烯树脂组合物的物性汇总显示于表1中。
实施例7
向设置有内容积10升的外壳、带搅拌机的反应器装入均匀混合了多官能乙烯基共聚物(α)0.06重量份的苯乙烯单体3kg、及包含作为悬浊稳定剂的磷酸三钙0.05重量份、作为表面活性剂的十二烷基苯磺酸钠0.005重量份的水4kg,在搅拌下使溶液悬浊。向该悬浊液添加相对于苯乙烯单体100重量份,作为聚合引发剂的叔丁基过氧苯甲酸酯0.2重量份,进一步添加作为链转移剂的α-甲基苯乙烯二聚物0.04重量份。一边搅拌悬浊液,一边在115℃时加热5小时,在140℃时加热3小时进行聚合。聚合结束之后,向悬浊液添加盐酸,中和作为悬浊稳定剂的磷酸三钙。将获得的珠状的树脂洗涤、过滤之后、进行热风干燥,获得苯乙烯系树脂组合物。
实施例8
除了代替实施例7中的多官能乙烯基共聚物(α),使用多官能乙烯基共聚物(β)以外,与实施例7同样地操作,获得苯乙烯系树脂组合物。
实施例9
除了代替实施例7中的多官能乙烯基共聚物(α),使用多官能乙烯基共聚物(γ)以外,与实施例7同样地操作,获得苯乙烯系树脂组合物。
实施例10
除了使实施例7中的多官能乙烯基共聚物(α)的添加量0.06重量份变为0.01重量份以外,与实施例7同样地操作,获得聚苯乙烯树脂组合物。
实施例11
除了使实施例7中的多官能乙烯基共聚物(α)的添加量0.06重量份变为0.1重量份以外,与实施例7同样地操作,获得聚苯乙烯树脂组合物。
比较例7
除了不添加多官能乙烯基共聚物(α)以外,与实施例7同样地操作,获得线状聚苯乙烯。
比较例8
除了使实施例7中的多官能乙烯基共聚物(α)的添加量0.06重量份变为0.001重量份以外,与实施例7同样地操作,获得聚苯乙烯树脂组合物。
比较例9
除了使实施例7中的多官能乙烯基共聚物(α)的添加量0.06重量份变为1重量份以外,与实施例7同样地操作,获得聚苯乙烯树脂组合物。
比较例10
除了代替实施例7中的多官能乙烯基共聚物(α),使二乙烯苯为0.05重量份以外,与实施例7同样地操作,获得聚苯乙烯树脂组合物。
将实施例7~11及比较例7~10中的反应原料的使用量及聚苯乙烯树脂组合物的物性汇总显示于表2中。
表1~2中,交联剂意指多官能乙烯基共聚物或二乙烯苯(DVB),链转移剂意指tDM。整体Mw意指聚苯乙烯树脂组合物的重均分子量,Mw1,000,000以上的比例意指聚苯乙烯树脂组合物中的比例(重量%),凝胶状物的评价中的Y意指存在凝胶状物,N意指不存在凝胶状物。
表1
表2
产业上利用的可能性
根据本发明,在伴随以发泡成形为代表的薄壁压延的加工中,可生产不包含诱发薄壁部的断裂的微凝胶,进一步,含有在以应变固化性为代表的溶融特性方面优异、压延时可均匀地壁厚化的高分支型超高分子量共聚物和线状聚合物的苯乙烯系树脂组合物。进一步,通过使用利用本发明而获得的苯乙烯系树脂组合物,抑制在片材成形中因二次加工时的下垂、厚度不均、凝胶状物导致的破裂、外观的恶化。另外,可消除在吹塑成形时的缩坯、发泡成形时的气泡破裂、气泡膨胀、连续地生成气泡等各种问题。
含有高分支型超高分子聚合物的苯乙烯系树脂组合物的制造方法及其组合物专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。















动态评分
0.0