








专利摘要
本发明公开了一种吩噻嗪基D‑A‑π‑A型有机染料的制备方法。主要采用二苯胺、吩噻嗪作为给电子单元,苯并噻二唑作为辅A单元,噻吩作为共轭桥连π单元,氰基丙烯酸作为吸电子A单元;通过C‑N偶联、NBS上溴、铃木偶联等合成方法制备D‑A‑π‑A型有机染料分子。本发明工艺简单,制得的吩噻嗪基D‑A‑π‑A型有机染料,其结构能有效拓宽染料吸收光谱,提高电子注入效率和能量转换效率,将其应用到染料敏化太阳能电池中可有效提高光电转化效率。
权利要求
1.一种吩噻嗪基D-A-π-A型有机染料的制备方法,其特征在于具体步骤为:
(1)量取100mL的分析纯四氢呋喃加入三口瓶中,称取10.0122g吩噻嗪和6.0334g分析纯叔丁基醇钾,惰性气体保护下,室温搅拌1小时,针头打入13.3mL分析纯溴代异辛烷,升温回流,柱色谱跟踪,反应物浓度不变,停止反应;后处理:冷却,旋干,分析纯二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,中性氧化铝柱色谱分离,分析纯石油醚洗脱,收集产物,真空干燥箱烘干至恒重,得10.0328g黄色油状液体即中间体1,产率64.6%;1H NMR(CDCl3,400MHz)δ(TMS,ppm):7.20-7.16(m,4H),7.14-7.08(m,2H),7.05-6.97(m,2H),2.40-2.28(t,1H),1.58-1.37(t,10H),0.88-0.72(m,6H).M/S=311.5;
(2)量取10mL的分析纯四氢呋喃加入三口瓶中,称取6.1923g中间体1冰水浴避光搅拌溶解,避光称取3.5600gN-溴代琥珀酰亚胺溶于50mL的分析纯四氢呋喃溶液中,避光恒压滴液漏斗缓慢滴加,柱色谱跟踪,反应物浓度不变,停止反应;后处理:冷却,旋干,分析纯二氯甲烷溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离,分析纯石油醚洗脱,收集产物,旋干得黄色油状液体,真空干燥箱烘干至恒重得中间体2,3.89g,产率50%;1HNMR(CDCl3,400MHz)δ(TMS,ppm):7.20-6.97(m,4H),6.82-6.80(m,3H),2.82-2.70(t,1H),2.43-2.25(t,1H),2.43-2.25(m,10H),0.88-0.71(m,6H).M/S=390.4;
(3)量取50mL的分析纯甲苯加入三口瓶中,称取3.07g中间体2、1.47g二苯胺和2.4680g分析纯叔丁基醇钠,搅拌溶解,惰性气体保护下,加入0.09g醋酸钯,升温回流,用薄层色谱跟踪反应至反应物浓度不变,停止反应;后处理:冷却,旋干,分析纯二氯甲烷溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离,分析纯二氯甲烷/分析纯石油醚,梯度洗脱,收集产物,旋干得淡绿色油状液体,真空干燥箱烘干至恒重得中间体3,1.9g,产率52%;1HNMR(CDCl3,400MHz)δ(TMS,ppm):7.23-7.18(m,6H),7.16-7.04(m,4H),6.97-6.87(m,4H),6.85-6.77(d,2H),1.80-1.62(t,1H),1.27-1.11(m,10H),0.88-0.74(m,6H).M/S=478.7;
(4)称取1.7263g中间体3,其他原料配比以及后处理与步骤(2)一致,得淡绿色黏稠液体即中间体4,0.8964g,产率44.5%;1H NMR(CDCl3,400MHz)δ(TMS,ppm):7.46-7.44(d,1H),7.26-7.19(m,5H),7.18-7.14(m,3H),6.97-6.83(d,5H),6.78-6.68(d,2H),2.15-2.06(m,1H),1.60-1.47(m,4H),1.40-1.32(m,2H),1.22-1.14(m,4H),0.85-0.74(m,6H).M/S=557.6;
(5)称取2.3860g的中间体4、1.6325g联硼酸频那醇酯和1.0553g乙酸钾,加入100mL的两口瓶中,量取50mL体积比为4:1的分析纯四氢呋喃和无水甲醇的混合溶液加入两口瓶中搅拌溶解,氩气保护,加入0.1050g[1,1'-双(二苯基膦基)二茂铁]二氯化钯,70℃升温回流,用薄层色谱跟踪反应至反应物浓度不变,停止反应;后处理:冷却,旋干,分析纯二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离,分析纯二氯甲烷/分析纯石油醚,梯度淋洗,收集产物,旋干得淡绿色固体即中间体5,1.2000g,产率55.8%;1H NMR(DMSO,500MHz)δ(TMS,ppm):7.75-7.63(d,5H),7.51-7.38(m,1H),7.35-7.24(m,3H),7.20-6.95(m,5H),6.85-6.50(d,2H),1.67-1.51(t,1H),1.46-1.38(m,10H),1.36-1.07(m,12H),0.80-0.75(m,6H).MS:m/z=501.4;
(6)量取90mL体积比为2:1的分析纯四氢呋喃和水的混合溶液加入两口瓶中,升温至沸,冷却至室温;称取1.617g 4,7-二溴-2、1,3-苯并噻二唑、0.7800g 5-醛基-2-噻吩硼酸和1.47g碳酸钾加入两口瓶中搅拌溶解,氩气保护;加入0.25g四三苯基磷钯和0.05mL分析纯三辛基甲基氯化铵,90℃回流,用薄层色谱跟踪反应至反应物浓度不变,停止反应;后处理:冷却,旋干,分析纯二氯甲烷溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离,分析纯乙酸乙酯/分析纯石油醚,梯度淋洗,收集产物,旋干得黄色固体,真空干燥箱70℃烘干至恒重得中间体6(0.4058g,产率25%);1H NMR(DMSO,500MHz),(DMSO,ppm):10.03(s,1H),8.30-8.27(d,1H),8.25-8.23(d,1H),8.17-8.13(t,2H).MS:m/z=325.1;
(7)称取0.7000g中间体5、0.1603g中间体6和0.3588g乙酸钾加入100mL的两口瓶中,加入33mL体积比为10:1的分析纯四氢呋喃和水的混合溶液搅拌溶解,氩气保护,称取0.0705g四三苯基磷钯加入两口瓶,氩气保护,升温回流,用薄层色谱跟踪反应至反应物浓度不变,停止反应;后处理:冷却,旋干,分析纯二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离,分析纯乙酸乙酯/分析纯石油醚,梯度淋洗,收集产物,旋干得紫黑色固体,真空干燥箱烘干至恒重得中间体7,0.1160g,产率32.3%;1H NMR(DMSO,500MHz)δ(TMS,ppm):8.75-8.70(s,1H),8.39-8.33(m,1H),8.31-8.23(m,2H),8.17-8.09(m,1H),8.06-7.88(d,3H),7.46-7.39(m,4H),7.31-7.15(m,3H),7.12-6.91(m,5H),3.83-3.42(m,2H),1.88-1.74(m,1H),1.47-1.33(m,4H),1.31-1.07(m,6H),0.88-0.75(m,6H).MS:m/z=723;
(8)量取20mL的分析纯氯仿加入三口瓶中,加入0.1160g中间体7和0.0833g氰基丙烯酸,搅拌溶解,惰性气体保护,打入0.0365g分析纯哌啶,升温回流,用薄层色谱跟踪反应至反应物浓度不变,停止反应;后处理:分析纯氯仿稀释,市售稀盐酸洗,水洗,收集有机相,旋干,硅胶柱色谱分离,分析纯二氯甲烷/无水甲醇,梯度淋洗,收集产物,旋干得紫黑色固体,真空干燥箱烘干至恒重得吩噻嗪基D-A-π-A型有机染料,0.0997g,产率78.9%;1H NMR(DMSO,500MHz)δ(TMS,ppm):8.61-8.52(s,1H),8.37-8.29(m,1H),8.28-8.20(m,2H),8.14-8.07(m,1H),8.01-7.86(d,3H),7.41-7.32(m,4H),7.27-7.10(m,3H),7.08-6.82(m,5H),3.81-3.43(m,2H),1.88-1.79(m,1H),1.49-1.36(m,4H),1.34-1.05(m,6H),0.91-0.71(m,6H).MS:m/z=790。
说明书
技术领域
本发明具体涉及一种D-A-π-A型吩噻嗪基染料的设计合成及其对光谱的响应。
技术背景
在染料敏化太阳能电池中,传统的有机染料设计主要遵循电子给体-共轭桥连单元-电子受体(D-π-A型)的思路,但其对设计宽光谱、高效率、高稳定性的有机敏化染料存在明显的局限性。近年来的研究是通过在电子给体D单元和共轭桥联π-单元之间引入一个额外的辅A单元,构筑D-A-π-A型染料分子。结果显示,这种新型的结构可有效拓宽染料吸收光谱,提高电子注入效率和能量转换效率。
吩噻嗪是一种强供电子单元,具有非平面的蝴蝶型分子结构,可有效的抑制染料分子聚集,减少暗电流,在染料的设计和合成中有着和广泛的应用。而苯并噻二唑是一类含有氮、硫杂环的化合物,由于具有较强的电子亲和势、共平面性和对化合物能级较好的可调性等特点,广泛应用于具有共轭结构的有机分子的构筑。
发明内容
本发明的目的是基于以上的经验,结合二苯胺、吩噻嗪和苯并噻二唑制备了一种D-A-π-A型有机染料。
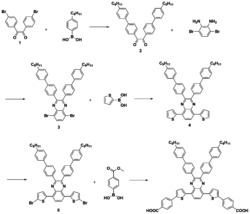
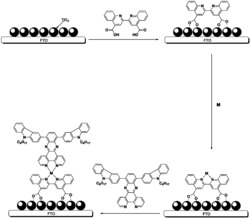
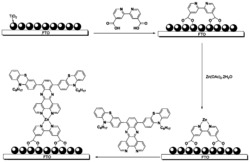
本发明涉及的吩噻嗪基D-A-π-A型有机染料的结构如图1,吩噻嗪基D-A-π-A型有机染料的具体合成路线见图2。
吩噻嗪基D-A-π-A型有机染料的制备方法具体步骤为:
(1)量取适量的四氢呋喃(THF)加入三口瓶中,称取1摩尔份吩噻嗪、1.1摩尔份叔丁基醇钾,惰性气体保护下,室温搅拌1小时,针头打入1.5摩尔份溴代异辛烷,升温回流,柱色谱跟踪,反应物浓度基本不变,停止反应。后处理:冷却,旋干,二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,中性氧化铝柱色谱分离(石油醚洗脱),收集产物,旋干得黄色油状液体,真空干燥箱烘干至恒重得中间体1。
(2)量取适量的THF加入三口瓶中,称取1摩尔份中间体1冰水浴避光搅拌溶解,避光称取1.1摩尔份NBS溶于适量的THF溶液中,恒压滴液漏斗滴加,柱色谱跟踪,反应物浓度基本不变,停止反应。后处理:冷却,旋干,二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离(石油醚洗脱),收集产物,旋干得黄色油状液体,真空干燥箱烘干至恒重得中间体2。
(3)量取适量的甲苯加入三口瓶中,称取1摩尔份中间体2、1.1摩尔份二苯胺、4摩尔份叔丁基醇钠,搅拌溶解,惰性气体保护下,加入0.05摩尔份醋酸钯,升温回流,用薄层色谱跟踪反应至反应物浓度基本不变,停止反应;后处理:冷却,旋干,二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离(二氯甲烷/石油醚,梯度洗脱),收集产物3,旋干得淡绿色油状液体,真空干燥箱烘干至恒重得中间体3。
(4)称取1摩尔份中间体3,其他原料配比以及后处理与步骤(2)一致,得淡绿色黏稠液体即中间体4。
(5)量取适量的THF加入两口瓶中,称取1摩尔份中间体4、0.5摩尔份联硼酸频那醇酯和0.3摩尔份的乙酸钾,搅拌溶解,惰性气体保护,加入0.05摩尔份[1,1'-双(二苯基膦基)二茂铁]二氯化钯,升温回流,用薄层色谱跟踪反应至反应物浓度基本不变,停止反应;后处理:冷却,旋干,二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离(二氯甲烷/石油醚,梯度淋洗),收集产物,旋干得淡绿色固体即中间体5.
(6)溶剂除氧:量取适量THF加入两口瓶中,升温至沸,冷却至室温;称取1摩尔份4,7-二溴-2,1,3-苯并噻二唑、0.5摩尔份5-醛基-2-噻吩硼酸、1份碳酸钾加入两口瓶中搅拌溶解,惰性气体保护,加入0.05摩尔份四三苯基磷钯,适量三辛基甲基氯化铵。高温回流,用薄层色谱跟踪反应至反应物浓度基本不变,停止反应;后处理:冷却,旋干,二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离,收集产物,旋干得黄色固体,烘干至恒重得中间体6.
(7)量取适量的THF加入两口瓶中,称取4摩尔份中间体5、1摩尔份中间体6和2摩尔份乙酸钾,搅拌溶解,惰性气体保护,称取0.05份四三苯基磷钯加入两口瓶,升温回流,用薄层色谱跟踪反应至反应物浓度基本不变,停止反应;后处理:冷却,旋干,二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离,收集产物,旋干得紫黑色固体,真空干燥箱烘干至恒重得中间体7。
(8)量取适量氯仿加入三口瓶中,称取1.2摩尔份中间体7,1摩尔份氰基丙烯酸,搅拌溶解,惰性气体保护,打入0.4份哌啶,升温回流,用薄层色谱跟踪反应至反应物浓度基本不变,停止反应;后处理:氯仿稀释,适量的稀盐酸洗,水洗,收集有机相,旋干,硅胶柱色谱分离,收集产物,旋干得紫黑色固体,真空干燥箱烘干至恒重得吩噻嗪基D-A-π-A型有机染料。
本发明工艺简单,制得的吩噻嗪基D-A-π-A型有机染料,其结构能有效拓宽染料吸收光谱,提高电子注入效率和能量转换效率,提高了光电转化效率。
附图说明
图1为本发明实施例吩噻嗪基D-A-π-A型有机染料PT5结构图。
图2为本发明实施例吩噻嗪基D-A-π-A型有机染料PT5染料分子的合成路线图,图中:i)3-溴代异辛烷,分析纯叔丁醇钾,分析纯四氢呋喃;ii)N-溴代琥珀酰亚胺,分析纯四氢呋喃;iii)二苯胺,乙酸钯,乙酸钾,分析纯四氢呋喃;iv)联硼酸频那醇酯,醋酸钾,[1,1'-双(二苯基膦基)二茂铁]二氯化钯,分析纯四氢呋喃;v)四三苯基磷钯,分析纯四氢呋喃;vi)分析纯哌啶,氰乙酸,分析纯氯仿)。
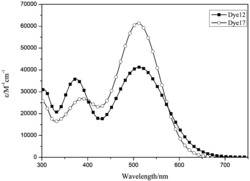
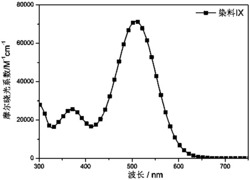
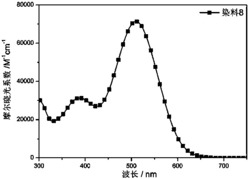
图3为本发明实施例吩噻嗪基D-A-π-A型有机染料PT5的紫外可见光谱图。
具体实施方式
实施例:
(1)中间体1的合成:量取100mL的分析纯四氢呋喃加入三口瓶中,称取10.0122g(50mmol)吩噻嗪和6.0334g(55mmol)分析纯叔丁基醇钾,惰性气体保护下,室温搅拌1小时,针头打入13.3mL(75mmol)分析纯溴代异辛烷,升温回流,柱色谱跟踪,反应物浓度基本不断,停止反应。后处理:冷却,旋干,分析纯二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,中性氧化铝柱色谱分离(分析纯石油醚洗脱),收集产物,真空干燥箱烘干至恒重,得10.0328g黄色油状液体即中间体1(产率64.6%)。1H NMR(CDCl3,400MHz)δ(TMS,ppm):7.20-7.16(m,4H),7.14-7.08(m,2H),7.05-6.97(m,2H),2.40-2.28(t,1H),1.58-1.37(t,10H),0.88-0.72(m,6H).M/S=311.5。
(2)中间体2的合成:量取10mL的分析纯四氢呋喃加入三口瓶中,称取6.1923g(20mmol)中间体1冰水浴避光搅拌溶解,避光称取3.5600g(20mmol)N-溴代琥珀酰亚胺溶于50mL的分析纯四氢呋喃溶液中,避光恒压滴液漏斗缓慢滴加,柱色谱跟踪,反应物浓度基本不断,停止反应。后处理:冷却,旋干,分析纯二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离(分析纯石油醚洗脱),收集产物,旋干得黄色油状液体,真空干燥箱烘干至恒重得中间体2(3.89g,产率50%)。1HNMR(CDCl3,400MHz)δ(TMS,ppm):7.20-6.97(m,4H),6.82-6.80(m,3H),2.82-2.70(t,1H),2.43-2.25(t,1H),2.43-2.25(m,10H),0.88-0.71(m,6H).M/S=390.4。
(3)中间体3的合成:量取50mL的分析纯甲苯加入三口瓶中,称取3.07g(7.9mmol)中间体2、1.47g(8.2mmol)二苯胺和2.4680g(24mmol)分析纯叔丁基醇钠,搅拌溶解,惰性气体保护下,加入0.09g(0.4mmol)醋酸钯,升温回流,用薄层色谱跟踪反应至反应物浓度基本不变,停止反应;后处理:冷却,旋干,分析纯二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离(分析纯二氯甲烷/分析纯石油醚,梯度洗脱),收集产物,旋干得淡绿色油状液体,真空干燥箱烘干至恒重得中间体3(1.9g,产率52%)。1H NMR(CDCl3,400MHz)δ(TMS,ppm):7.23-7.18(m,6H),7.16-7.04(m,4H),6.97-6.87(m,4H),6.85-6.77(d,2H),1.80-1.62(t,1H),1.27-1.11(m,10H),0.88-0.74(m,6H).M/S=478.7。
(4)中间体4的合成:称取1.7263g(3.614mmol)中间体3,其他原料配比以及后处理与步骤(2)一致,得淡绿色黏稠液体即中间体4(0.8964g,产率44.5%)。1H NMR(CDCl3,400MHz)δ(TMS,ppm):7.46-7.44(d,1H),7.26-7.19(m,5H),7.18-7.14(m,3H),6.97-6.83(d,5H),6.78-6.68(d,2H),2.15-2.06(m,1H),1.60-1.47(m,4H),1.40-1.32(m,2H),1.22-1.14(m,4H),0.85-0.74(m,6H).M/S=557.6。
(5)中间体5的合成:称取2.3860g(4.29mmol)的中间体4、1.6325g(6.43mmol)联硼酸频那醇酯和1.0553g(12.87mmol)乙酸钾,加入100mL的两口瓶中,量取50mL体积比为4:1的分析纯四氢呋喃和无水甲醇的混合溶液加入两口瓶中搅拌溶解,氩气保护,加入0.1050g(0.13mmol)[1,1'-双(二苯基膦基)二茂铁]二氯化钯,70℃升温回流,用薄层色谱跟踪反应至反应物浓度基本不变,停止反应;后处理:冷却,旋干,分析纯二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离(分析纯二氯甲烷/分析纯石油醚,梯度淋洗),收集产物,旋干得淡绿色固体即中间体5(1.2000g,产率55.8%)。1H NMR(DMSO,500MHz)δ(TMS,ppm):7.75-7.63(d,5H),7.51-7.38(m,1H),7.35-7.24(m,3H),7.20-6.95(m,5H),6.85-6.50(d,2H),1.67-1.51(t,1H),1.46-1.38(m,10H),1.36-1.07(m,12H),0.80-0.75(m,6H).MS:m/z=501.4。
(6)中间体6的合成:溶剂除氧:量取90mL(分析纯四氢呋喃/水,2/1,v/v)加入两口瓶中,升温至沸,冷却至室温;称取1.617g(5.5mmol)4,7-二溴-2、1,3-苯并噻二唑、0.7800g(5mmol)5-醛基-2-噻吩硼酸和1.47g(40mmol)碳酸钾加入两口瓶中搅拌溶解,氩气保护;加入0.25g(0.25mmol)四三苯基磷钯和0.05mL分析纯三辛基甲基氯化铵,90℃回流,用薄层色谱跟踪反应至反应物浓度基本不变,停止反应;后处理:冷却,旋干,分析纯二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离(分析纯乙酸乙酯/分析纯石油醚,梯度淋洗),收集产物,旋干得黄色固体,真空干燥箱70℃烘干至恒重得中间体6(0.4058g,产率25%)。1H NMR(DMSO,500MHz),(DMSO,ppm):10.03(s,1H),8.30-8.27(d,1H),8.25-8.23(d,1H),8.17-8.13(t,2H).MS:m/z=325.1。
(7)中间体7的合成:称取0.7000g(1.16mmol)中间体5、0.1603g(0.49mmol)中间体6和0.3588g(3.66mmol)乙酸钾加入100mL的两口瓶中,加入33mL体积比为10:1的分析纯四氢呋喃和水的混合溶液搅拌溶解,氩气保护,称取0.0705g(0.061mmol)四三苯基磷钯加入两口瓶,氩气保护,升温回流,用薄层色谱跟踪反应至反应物浓度基本不变,停止反应;后处理:冷却,旋干,分析纯二氯甲烷(DCM)溶解,水洗,萃取,收集有机相,旋干,硅胶柱色谱分离(分析纯乙酸乙酯/分析纯石油醚,梯度淋洗),收集产物,旋干得紫黑色固体,真空干燥箱烘干至恒重得中间体7(0.1160g,产率32.3%)。1H NMR(DMSO,500MHz)δ(TMS,ppm):8.75-8.70(s,1H),8.39-8.33(m,1H),8.31-8.23(m,2H),8.17-8.09(m,1H),8.06-7.88(d,3H),7.46-7.39(m,4H),7.31-7.15(m,3H),7.12-6.91(m,5H),3.83-3.42(m,2H),1.88-1.74(m,1H),1.47-1.33(m,4H),1.31-1.07(m,6H),0.88-0.75(m,6H).MS:m/z=723。
(8)染料PT5的合成(染料分子PTZ-5的合成):量取20mL的分析纯氯仿加入三口瓶中,加入0.1160g(0.16mmol)中间体7和0.0833g(0.98mmol)氰基丙烯酸,搅拌溶解,惰性气体保护,打入0.0365g(0.43mmol)分析纯哌啶,升温回流,用薄层色谱跟踪反应至反应物浓度基本不变,停止反应;后处理:分析纯氯仿稀释,市售稀盐酸洗,水洗,收集有机相,旋干,硅胶柱色谱分离(分析纯二氯甲烷/无水甲醇,梯度淋洗),收集产物,旋干得紫黑色固体,真空干燥箱烘干至恒重得染料PT5(0.0997g,产率78.9%)。1H NMR(DMSO,500MHz)δ(TMS,ppm):8.61-8.52(s,1H),8.37-8.29(m,1H),8.28-8.20(m,2H),8.14-8.07(m,1H),8.01-7.86(d,3H),7.41-7.32(m,4H),7.27-7.10(m,3H),7.08-6.82(m,5H),3.81-3.43(m,2H),1.88-1.79(m,1H),1.49-1.36(m,4H),1.34-1.05(m,6H),0.91-0.71(m,6H).MS:m/z=790。
图3紫外光谱图可以看出最大吸收波长达到了506nm,带隙宽度(E0-0)2.28eV,能有效地对太阳光进行吸收。
一种吩噻嗪基D-A-π-A型有机染料的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











动态评分
0.0