

IPC分类号 : C07J7/00,C07J9/00,C07J1/00,C07J13/00,C07J71/00,C07J5/00,C07J11/00,C07J75/00,A61K31/57,A61K31/575,A61K31/568,A61K31/56,A61K31/5685,A61K31/58,A61K31/573,A61P3/04,A61P3/10,A61P3/06,A61P9/12,A61P9/00,A61P25/18,A61P25/28,A61P5/00




专利摘要
式(I)所示的达玛烷型三萜衍生物及其药用盐,其制备方法,以其为有效成分的药物组合物,它们在制备治疗或预防肥胖、糖尿病、高血压及心脑血管疾病及其相关的代谢性疾病药物中的应用,它们在制备11β-HSD1抑制剂中的应用。
权利要求
1.式(I)所示的达玛烷型三萜衍生物或其药用盐,
式中小写字母a~e代表独立的双键或单键,用 表示;
其中a为双键时,R1、R2相同或不同,分别选自H或R1选自H,R2选自=O、=S、-OH、卤素、C1-10烷氧基、C1-10酯基、-NH2/-NR2,其中R为H和C1-10烷基或都为C1-10烷基;a为单键时,R1选自H,R2选自=O、=S、-OH、卤素、C1-10烷氧基、C1-10酯基;
a~e为单键或双键时,R3选自H、=O、=S、-OH、卤素、C1-10烷氧基、C1-10酯基、-NH2/-NR2,其中R为H和C1-10烷基或都为C1-10烷基;
b为双键时,R4选自H;b为单键时,R4选自=O、=S、-OH、卤素、C1-10烷氧基、C1-10酯基、-NH2/-NR2,其中R为H和C1-10烷基或都为C1-10烷基;
c、d分别为双键时,R5=R6选自H或甲基;除此之外,R5=-OH、H,R6为-COCHO、-COCOOH、-COCH2R、-COCOR,其中R为H、-OH、-NH2、卤素、C1-10烷基、C2-10烯基、C1-10烷氧基、C1-10酯基、6元芳基、5至6元杂芳基、C1-10酰胺基、C3-C10含N原子环烷基、C1-10烷基胺基、相同与不同的C1-10二烷基胺基;或R5 R6 =O、H;
d、e分别为双键时,R7选自H;d、e都为单键时,R7选自H、卤素、C1-10烷基、C2-10烯基;
e为双键时,R8选自H或甲基;e为单键时,R8选自H或C1-10烷基、C2-10烯基、6元芳基、5至6元杂芳基、-NR2,其中R为H和C1-10烷基或都为C1-10烷基。
2.如权利要求1式(I)所示的达玛烷型三萜衍生物,为如下式(II)所示的达玛烷型三萜衍生物,
其中R1、R2相同或不同,分别选自-OH、=O、-OCH2OCH3、-OTBS、-OTBDPS、-OBn、-OCOR,其中R为C1-10烷基;
a为双键时,R3=R4选自H或甲基;a为单键时,R3=-OH、H, R4为-COCHO、-COCOOH、-COCH2R、-COCOR,其中R为H、-OH、-NH2、卤素、C1-10烷基、C2-10烯基、C1-10烷氧基、C1-10酯基、6元芳基、5至6元杂芳基、C1-10酰胺基、C3-C10含N原子环烷基、C1-10烷基胺基、相同与不同的C1-10二烷基胺基;或R3 R4 =O、H;
R5选自H,=O。
3.如权利要求1或2所示的达玛烷型三萜衍生物,为如下化合物:
4.权利要求1或2或3所示的达玛烷型三萜衍生物的制备方法,包括下述步骤:
(1)将20(S)/20(R)-原人参二醇(R3=H)或20(S)/20(R)-原人参三醇(R3=OH)化合物7催化氢化,然后用乙酰氧基保护二级羟基,得化合物8,然后将化合物8消除和氧化断裂转化成化合物1,化合物1换MOMO保护得到化合物9,化合物9通过还原、消除及氧化裂解得到化合物10,化合物10经氧化、还原及环氧化得到化合物11,化合物11经甲基化与环氧开环得到化合物12,化合物12经氧化、去保护及选择性保护,得到化合物3;
(2)将化合物8用三氯氧磷/吡啶处理后,接着与三氯化钌/高碘酸钠反应,得化合物2,
(3)将化合物3用碳酸钾/甲醇处理后,得化合物4,
(4)将化合物3用DBU处理后,得化合物6,
(5)将化合物6用碳酸钾/甲醇处理后,得化合物5,
。
5.用于治疗或预防肥胖及与其相关的代谢性疾病的药物组合物,其中含有治疗有效量的权利要求1或2或3所示的达玛烷型三萜衍生物或其药用盐和药学上可接受的载体。
6.11β-HSD1抑制剂,其中含有治疗有效量的权利要求1或2或3所示的达玛烷型三萜衍生物或其药用盐和药学上可接受的载体。
7.如权利要求1所述的所示的达玛烷型三萜衍生物或其药用盐,其特征在于所述的药用盐是指药学上可接受的盐,是与有机酸形成的盐,所述的有机酸为酒石酸、柠檬酸、甲酸、乙酸、乙二酸、丁酸、草酸、马来酸、琥珀酸、己二酸、藻酸、柠檬酸、天冬氨酸、苯苯磺酸、樟脑酸、樟脑磺酸、二葡糖酸、环戊烷丙酸、十二烷基硫酸、乙磺酸、葡庚糖酸、甘油磷酸、半硫酸、庚酸、己酸、延胡索酸、2-羟基乙磺酸、乳酸、马来酸、甲磺酸、烟酸、2-萘磺酸、扑酸、果胶酯酸、3-苯基丙酸、苦味酸、新戊酸、丙酸、琥珀酸、酒石酸、硫代氰酸、对-甲苯磺酸盐和十一烷酸盐。
8.权利要求1或2或3所示的达玛烷型三萜衍生物或其药用盐在制备治疗或预防肥胖及与其相关的代谢性疾病的药物中的应用。
9.如权利要求8所述的应用,其中所述的肥胖及与其相关的代谢性疾病为2型糖尿病、血脂代谢障碍、高血压及心血管并发症、精神性疾病、认知障碍、神经退行性病变、神经内分泌失调。
10.权利要求1或2或3所述的达玛烷型三萜衍生物或其药用盐在制备11β-HSD1抑制剂中的应用。
说明书
技术领域:
本发明属于药物技术领域,具体地,涉及达玛烷三萜衍生物,其制备方法以及其药物组合物在制备治疗或预防肥胖、糖尿病、高血压及心脑血管疾病及其相关的代谢性疾病药物中的应用。
背景技术:
肥胖及与其相关的代谢性疾病(包括2型糖尿病,血脂代谢障碍,高血压及心血管并发症等)已成为困扰现代人的主要健康问题。据世界健康组织(WHO)不完全统计显示,全球约有16亿人体重超重,其中有4亿人表现出临床的糖尿病症状,且情况还在进一步恶化之中。尽管目前已发现多个靶标与代谢综合症相关,如过氧化物酶增殖体活化受体 (Peroxi- some proliferators activated receptors, PPARs) α、β、δ、γ, 11β-羟化类固醇脱氢酶1型酶 (11β- HSD1)(Schwenger, P., Bellosta, P., Vietor, I., Basilico, C., Skolnik, E. Y., Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 2869)、肝X受体 (LXR)、大麻素受体1 (CB1)等,并创制了一些有效的药物,但与紧迫的形式相比仍然相形见绌。因此,严峻的形势赋予了医药工作者责无旁贷的使命——寻找新型高效低副作用的糖尿病及其并发症的治疗药物。
鉴于11β-HSD1是调节糖皮质激素代谢的一个关键酶,而且越来越多的证据已经表明11β-HSD1 抑制剂能有效的治疗代谢综合症。由于11β-HSD1在糖皮质激素水平调节中的重要作用(Kannisto, K., Pietilainen, K. H., Ehrenhorg, E., Rissanen, A., Kaprio, J., Hamsten, A., Yki-Jarvien, H., J. Clin. Endocrinol. Metab., 2004, 89, 4414)(Masuzaki, H., Paterson, J., Shinyama, H., Morton, N. M., Mullins, J. J., Seckl, J. R., Flier, J. S., Science, 2001, 294, 2166),众多大型医药公司和研究机构都将目光集中于11β-HSD1,研究开发11β-HSD1选择性抑制剂。自Biovitrum公司报道了第一个选择性11β-HSD1抑制剂BVT- 2733以来,已有众多11β-HSD1选择性抑制剂被报道。
此外,目前进入临床的11β-HSD1抑制剂均是合成产物,尚未有源自天然的11β-HSD1抑制剂进入临床研究的报道。而天然产物不仅是一个很大的结构多样性库,同时也是一个功能多样的化合物资源库。之前本申请人曾从天然产物中发现了两类新型的11β-HSD1选择性抑制剂药物先导化合物(中国专利申请“丹参酮衍生物及其药物组合物和其在医药中的用途”,公开号CN 102304166 A)(中国专利申请“贝壳杉烷型二萜衍生物及其药物组合物和其在医药中的用途”,申请号201210044133.X)。迄今,现有技术中未见有达玛烷型三萜化合物作为11β-HSD1抑制剂的报道。
发明内容:
本发明的目的在于:提供达玛烷型三萜衍生物,其制备方法;以及达玛烷型三萜衍生物及其药物组合物在制备预防或治疗肥胖、糖尿病、高血压及心脑血管疾病及其相关的代谢性疾病的药物中的应用。
本发明的上述目的是通过如下的技术方案得以实现的:
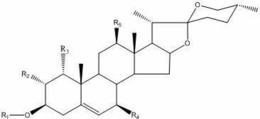
式(I)所示的达玛烷型三萜衍生物或其药用盐,
式中小写字母a~e代表独立的双键或单键,用 表示;
其中a为双键时,R1、R2相同或不同,分别选自H或R1选自H,R2选自=O、=S、-OH、卤素、C1-10烷氧基、C1-10酯基、-NH2/-NR2,其中R为H和C1-10烷基或都为C1-10烷基;a为单键时,R1选自H,R2选自=O、=S、-OH、卤素、C1-10烷氧基、C1-10酯基;
a~e为单键或双键时,R3选自H、=O、=S、-OH、卤素、C1-10烷氧基、C1-10酯基、-NH2/-NR2,其中R为H和C1-10烷基或都为C1-10烷基;
b为双键时,R4选自H;b为单键时,R4选自=O、=S、-OH、卤素、C1-10烷氧基、C1-10酯基、-NH2/-NR2,其中R为H和C1-10烷基或都为C1-10烷基;
c、d分别为双键时,R5=R6选自H或甲基;除此之外,R5=-OH、H,R6为-COCHO、-COCOOH、-COCH2R、-COCOR,其中R为H、-OH、-NH2、卤素、C1-10烷基、C2-10烯基、C1-10烷氧基、C1-10酯基、6元芳基、5至6元杂芳基、C1-10酰胺基、C3-C10含N原子环烷基、C1-10烷基胺基、相同与不同的C1-10二烷基胺基;或R5 R6 =O、H;
d、e分别为双键时,R7选自H;d、e都为单键时,R7选自H、卤素、C1-10烷基、C2-10烯基;
e为双键时,R8选自H或甲基;e为单键时,R8选自H或C1-10烷基、C2-10烯基、6元芳基、5至6元杂芳基、-NR2,其中R为H和C1-10烷基或都为C1-10烷基。
上述的达玛烷型三萜衍生物,优选如下式(II)所示的达玛烷型三萜衍生物,
其中R1、R2相同或不同,分别选自-OH、=O、-OCH2OCH3、-OTBS、-OTBDPS、-OBn、-OCOR,其中R为C1-10烷基;
a为双键时,R3=R4选自H或甲基;a为单键时,R3=-OH、H, R4为-COCHO、-COCOOH、-COCH2R、-COCOR,其中R为H、-OH、-NH2、卤素、C1-10烷基、C2-10烯基、C1-10烷氧基、C1-10酯基、6元芳基、5至6元杂芳基、C1-10酰胺基、C3-C10含N原子环烷基、C1-10烷基胺基、相同与不同的C1-10二烷基胺基;或R3 R4 =O、H;
R5选自H,=O。
上述的达玛烷型三萜衍生物,优选如下化合物:
本发明的达玛烷型三萜衍生物的制备方法,包括下述步骤:
(1)将20(S)/20(R)-原人参二醇(R3=H)或20(S)/20(R)-原人参三醇(R3=OH)化合物7催化氢化,然后用乙酰氧基保护二级羟基,得化合物8,然后将化合物8消除和氧化断裂转化成化合物1,化合物1换MOMO保护得到化合物9,化合物9通过还原、消除及氧化裂解得到化合物10,化合物10经氧化、还原及环氧化得到化合物11,化合物11经甲基化与环氧开环得到化合物12,化合物12经氧化、去保护及选择性保护,得到化合物3;
(2)将化合物8用三氯氧磷/吡啶处理后,接着与三氯化钌/高碘酸钠反应,得化合物2,
(3)将化合物3用碳酸钾/甲醇处理后,得化合物4,
(4)将化合物3用DBU处理后,得化合物6,
(5)将化合物6用碳酸钾/甲醇处理后,得化合物5,
。
用于治疗或预防肥胖及与其相关的代谢性疾病的药物组合物,其中含有治疗有效量的上述所示的达玛烷型三萜衍生物或其药用盐和药学上可接受的载体。
11β-HSD1抑制剂,其中含有治疗有效量的上述所示的达玛烷型三萜衍生物或其药用盐和药学上可接受的载体。
本发明所述的药用盐是指药学上可接受的盐,是与有机酸形成的盐,所述的有机酸为酒石酸、柠檬酸、甲酸、乙酸、乙二酸、丁酸、草酸、马来酸、琥珀酸、己二酸、藻酸、柠檬酸、天冬氨酸、苯苯磺酸、樟脑酸、樟脑磺酸、二葡糖酸、环戊烷丙酸、十二烷基硫酸、乙磺酸、葡庚糖酸、甘油磷酸、半硫酸、庚酸、己酸、延胡索酸、2-羟基乙磺酸、乳酸、马来酸、甲磺酸、烟酸、2-萘磺酸、扑酸、果胶酯酸、3-苯基丙酸、苦味酸、新戊酸、丙酸、琥珀酸、酒石酸、硫代氰酸、对-甲苯磺酸盐和十一烷酸盐。
本发明的达玛烷型三萜衍生物或其药用盐在制备治疗或预防肥胖及与其相关的代谢性疾病的药物中的应用。
所述的应用,其中肥胖及与其相关的代谢性疾病为2型糖尿病、血脂代谢障碍、高血压及心血管并发症、精神性疾病、认知障碍、神经退行性病变、神经内分泌失调。
本发明还提供了达玛烷型三萜衍生物或其药用盐在制备11β-HSD1抑制剂中的应用。
除非另有说明,本发明使用的术语“烷基” 是指直链或者支链的饱和一价烃基,其中烷基可以任选地被一个或多个取代基取代。术语“烷基”也包括直链和支链烷基,除非另作说明。在某些实施方式中,烷基是具有1到20(C1-20)、1到15(C1-15)、1到12(C1-12)、1到10(C1-10)、或者1到6(C1-6)个碳原子的直链饱和的一价烃基,或者具有3到20(C3-20)、3到15(C3-15)、3到12(C3-12)、3到10(C3-10)、或者3到6(C3-6)个碳原子的支链的饱和一价烃基。本发明使用的直链C1-6 和支链C3-6烷基也被称为“低级烷基”。烷基的实施方式包括但不限于甲基、乙基、丙基(包括所有同分异构形式)、n-丙基、异丙基、丁基(包括所有同分异构形式)、n-丁基、异丁基、t-丁基、戊基(包括所有同分异构形式)、和己基(包括所有同分异构形式)。例如,C1-6烷基指具有1到6个碳原子的直链饱和一价烃基或者具有3到6个碳原子的支链饱和一价烃基。
除非另作说明,本发明使用的术语“烯基”是指包含一个或多个碳碳双键(在一个实施方式中1 至5个)的直链或者支链一价烃基。烯基可以任选地被一个或多个取代基取代。术语“烯基”也包括 “顺式(cis)”和“反式(trans)”结构的基团,或者本领域普通技术人员所理解的“E”和“Z”式结构。除非另作说明,本发明使用的术语“烯基”包括直链和支链烯基。例如,C2-6烯基是指2到6个碳原子的直链不饱和一价烃基或者3到6个碳原子的支链不饱和一价烃基。在某些实施方式中,烯基是2到20(C2-20)、2到15(C2-15)、2到12(C2-12)、2到10(C2-10)、或者2到6(C2-6)个碳原子的直链一价烃基,或者3到20(C3-20)、3到15(C3-15)、3到12(C3-12)、3到10(C3-10)、或者3到6(C3-6)个碳原子的支链一价烃基。烯基的实施方式包括但不限于乙烯基、丙烯-1-基、丙烯-2-基、烯丙基、丁烯基、以及4-甲基丁烯基。
除非另作说明,本发明使用的术语“炔基”是指包含一个或多个碳碳三键(在一个实施方式中1 到5个)的直链或者支链一价烃基。炔基可以任选地被一个或多个取代基取代。除非另作说明,术语“炔基”也包括直链和支链炔基。在某些实施方式中,炔基是2到20(C2-20)、2到15(C2-15)、2到12(C2-12)、2到10(C2-10)、或者2到6(C2-6)个碳原子的直链一价烃基,或者3到20(C3-20)、3到15(C3-15)、3到12(C3-12)、3到10(C3-10)、或者3到6(C3-6)个碳原子的支链的一价烃基。炔基的实施方式包括但不限于乙炔基(-C≡CH)和炔丙基(-CH2C≡CH)。例如,C2-6炔基指2到6个碳原子的直链不饱和一价烃基或者3到6个碳原子的支链不饱和一价烃基。
除非另作说明,本发明使用的术语“环烷基”是指环状饱和桥和/或非桥的一价烃基,其可以任选地被一个或多个本发明所述的取代基取代。在某些实施方式中,环烷基具有从3到20(C3-20)、从3到15(C3-15)、从3到12(C3-12)、从3到10(C3-10)、或者从3到7(C3-7)个碳原子。环烷烃基的实施方式包括但不限于环丙基、环丁基、环戊基、环己基、环庚基、十氢萘基和金刚烷基。
除非另作说明,本发明使用的术语“杂烷基”、“杂烯基”和“杂炔基”分别是指一个或多个碳原子被杂原子替换的烷基、链烯基和炔基。
除非另作说明,本发明使用的术语“杂原子”是指除了碳或者氢以外的其他任何原子。在某些实施方式中,术语“杂原子”是指N、O、S、Si或P。在其他实施方式中,术语“杂原子”是指N、O或者S。
除非另作说明,本发明使用的术语“芳基”是指单环芳基和/或包含至少一个芳香烃环的多环单价芳基。在某些实施方式中,芳基具有从6到20(C6-20)、从6到15(C6-15)、或者从6到10(C6-10)个环原子。芳基的实施方式包括但不限于苯基、萘基、芴基、薁基(azulenyl)、蒽基、菲基、芘基、联苯基和三联苯基。芳基也指其中一个环是芳香族的和其他环可以是饱和的,部分不饱和的或者芳香族的二环或者三环的碳环,例如二氢萘基、茚基、二氢茚基或者四氢萘基(萘满基(tetralinyl))。在某些实施方式中,芳基也可以任选地被一个或多个取代基取代。
除非另作说明,本发明使用的术语“芳基烷基”或者“芳烷基”是指用芳基取代的单价烷基。在某些实施方式中,烷基和芳基可任选地被一个或多个取代基取代。
本发明的化合物降达玛烷型三萜衍生物或其盐可经口或不经过口给药,给药量因药物不同而各有不同,对成人来说,每天1-1000mg比较合适。
具体实施方式:
下面的实施例可更详细地说明本发明,但不以任何形式限制本发明。
实施例1:
本发明的化合物达玛烷型三萜衍生物具有明显的11β-HSD1抑制活性,实验方法和结果如下:
一、材料与方法:
1.样品及制备:
样品呈无色,二甲基亚砜(DMSO)溶解配制为10mg/ml浓度的贮存液避光保存备用。
2.生物学模型11β-HSD1:
人源11β-HSD1;鼠源11β-HSD1。
3.实验方法:
(1)采用SPA(Scintillation proximity assay, 液闪接近测定技术)方法,对所有衍生物进行了对鼠源11β-HSD1和人源11β-HSD1 抑制作用的初筛,选择1μM 作为初筛浓度,对初筛抑制率超过50%的衍生物进行复筛,开展量效关系研究,并计算IC50值。
(2)阳性对照:甘草次酸(人源11β-HSD1和鼠源11β-HSD1)
二、结果:
表1 达玛烷三萜衍生物对人源及鼠源11β-HSD1的半数抑制浓度(IC50, μM)
注:ND为未测定
试验结果显示:本发明对一系列达玛烷型三萜衍生物进行了体外人源、鼠源11β-HSD1抑制活性评价,结果显示该类化合物对人源、鼠源11β-HSD1具有显著地抑制活性,且有着较好的选择性(30~2660)。
实施例2:
化合物8的制备:
将20 g 20(s)-原人参二醇(化合物7)溶于450 ml甲醇,加入适量的Raney Ni,接上氢气球,抽排三次除去体系中的空气,室温搅拌过夜,硅藻土过滤,收集滤液,减压蒸除溶剂,得到白色固体化合物13。 ESI: 485 [M+23]+, 1H-NMR( CDCl3,500MHz) 特征信号 δ:3.51-3.61(1H, td, J=5.2/10.3/10.3, H-C(12)),3.17-3.21(1H, dd, J=4.9/11.2, H-C(3)),1.99-2.06(1H, td, J=7.1/10.6/10.6, H-C(13))。
将5.7 g 化合物13溶于120 ml二氯甲烷,加入10.27ml三乙胺置于0 oC下搅拌10min, 0 oC下缓慢滴加4.66ml Ac2O的二氯甲烷溶液80 ml,滴完后室温下搅拌4h,用饱和NaCl溶液洗涤3次,有机相用无水NaSO4干燥,减压蒸除溶剂,得到淡黄色固体化合物8。 ESI: 569 [M+23]+, 1H-NMR( CDCl3,500MHz) 特征信号 δ:4.68-4.75(1H, td, J=4.7/10.1/10.1, H-C(12)),4.45-4.49(1H, dd, J=4.4/11.2, H-C(3))。
实施例3:
化合物1、2的制备:
将15 g 化合物8溶于150 ml吡啶,0 oC下缓慢滴加4 eq POCl3的吡啶溶液50 ml,滴完后40 oC下搅拌8h,乙酸乙酯稀释,用饱和CuSO4溶液洗涤3次,有机相用无水NaSO4干燥,减压蒸除溶剂,得到淡黄色固体化合物14。 ESI: 551 [M+23]+, 1H-NMR( CDCl3,500MHz) 特征信号 δ:5.03-5.06(1H, t, J=6.6, H-C(22))4.89-4.96(1H, td, J=5.3/10.9/10.9, H-C(12)),4.45-4.49(1H, dd, J=4.7/11.2, H-C(3)),2.44-2.57(1H, m, H-C(17))。
将4 g 化合物14溶于40 ml CCl4,加入60 ml饱和NaHCO3溶液与8.09 g NaIO4,搅拌15 分钟, 加入0.198 g RuCl3·nH2O的MeCN溶液40 ml,上述混合物在室温下避光搅拌4天, 硅藻土过滤,加入100 ml乙酸乙酯,用饱和NaCl溶液洗涤3次,有机相用无水Na2SO4干燥,减压蒸除溶剂,得到黑色油状物,该油状物经硅胶柱层析(石油醚:乙酸乙酯=10:1)得到化合物1(产率68%)为白色针状晶体。 ESI: 463 [M+23]+, 1H-NMR( CDCl3,500MHz) 特征信号 δ:4.74-4.81(1H, td, J=5.2/11/11, H-C(12)),4.46-4.50(1H, dd, J=4.7/11.3, H-C(3)),2.84-2.91(1H, td, J=6.1/10.9/10.9, H-C(17)),2.32(1H, t, J=11.0, H-C(13))。化合物2(产率22%)为白色片状晶体。
实施例4:
化合物9的制备:
将5 g 化合物1溶于100 ml甲醇,加入2.5eq K2CO3,滴完后40 oC下搅拌4h,减压蒸除溶剂,得到淡黄色固体。将该黄色固体溶于60 ml 二氯甲烷,0 °C下加入7.6eq DIPEA与MOMOCl,搅拌15 分钟, 室温搅拌2h,加入100 ml乙酸乙酯,用饱和NaCl溶液洗涤3次,有机相用无水Na2SO4干燥,减压蒸除溶剂,得到黄色油状物,该油状物经硅胶柱层析(石油醚:乙酸乙酯=30:1)得到化合物9为淡黄色油状物。 ESI: 487 [M+23]+, 1H NMR (CDCl3, 400 MHz) 特征信号δ 4.97 – 4.38 (m, 4H), 3.50 – 3.23 (m, 6H), 3.10 – 2.99 (m, 1H), 2.79 – 2.61 (m, 1H), 2.18 (dt, J = 20.4, 8.9 Hz, 3H), 2.06 – 1.88 (m, 3H), 1.79 – 1.63 (m, 4H), 1.63 – 1.48 (m, 5H), 1.02 (d, J = 12.6 Hz, 3H), 0.93 (d, J = 10.9 Hz, 4H), 0.79 (d, J = 7.9 Hz, 3H)。
实施例5:
化合物10的制备:
将5 g 化合物9溶于100 ml四氢呋喃,0 oC下分3次缓慢加入2eq LiAlH4,滴完后室温下搅拌1h,0 oC下滴加1 ml水,3 ml 15% NaOH溶液,搅拌,过滤,滤饼用乙酸乙酯洗3次,减压蒸除溶剂,得到淡黄色固体。将该黄色固体溶于60 ml 甲苯,加入3eq Burgess试剂,搅拌15 分钟, 70 oC搅拌8h,加入10 ml饱和NH4Cl溶液和50 ml乙酸乙酯搅拌分层,有机相用饱和NaCl溶液洗涤3次,无水Na2SO4干燥,减压蒸除溶剂,得到黄褐色油状物,该油状物经硅胶柱层析(石油醚:乙酸乙酯=30:1)得到淡黄色油状物。 将2 g 该淡黄色油状物溶于20 ml CCl4,加入30 ml饱和NaHCO3溶液与10eq NaIO4,搅拌15 分钟, 加入0.05eq RuCl3·nH2O的MeCN溶液20 ml,上述混合物在室温下避光搅拌4天, 硅藻土过滤,加入60 ml乙酸乙酯,用饱和NaCl溶液洗涤3次,有机相用无水Na2SO4干燥,减压蒸除溶剂,得到黑色油状物,该油状物经硅胶柱层析(石油醚:乙酸乙酯=11:1)得到化合物10为无色油状物。 ESI: 459 [M+23]+, 1H NMR (CDCl3, 600 MHz) 特征信号δ 4.96 (d, J = 7.0 Hz, 1H), 4.73 (dd, J = 17.1, 6.9 Hz, 2H), 4.64 – 4.58 (m, 1H), 3.75 – 3.65 (m, 1H), 3.45 (s, 3H), 3.39 (s, 3H), 3.13 – 3.05 (m, 1H), 2.32 (t, J = 13.1 Hz, 1H), 2.24 (ddd, J = 13.9, 12.0, 6.9 Hz, 1H), 2.19 – 2.10 (m, 1H), 2.07 – 1.97 (m, 1H), 1.96 – 1.86 (m, 1H), 1.80 – 1.67 (m, 4H), 1.67 – 1.55 (m, 3H), 1.52 – 1.38 (m, 6H), 1.31 – 1.18 (m, 3H), 1.11 – 1.06 (m, 4H), 1.00 – 0.95 (m, 4H), 0.94 – 0.84 (m, 10H), 0.82 – 0.78 (m, 4H).。
实施例6:
化合物11的制备:
将1 g 化合物10溶于20 ml四氢呋喃,-78 oC下分3次缓慢加入1.2eq LiHMDS,滴完后在该温度下搅拌1h,-78 oC下滴加1.2eq PhSeBr的四氢呋喃溶液10 ml,-78 oC搅拌反应2h,加入2 ml饱和NH4Cl溶液20 ml乙酸乙酯搅拌分层,有机相用饱和NaCl溶液洗涤3次,无水Na2SO4干燥,减压蒸除溶剂,得到黄色油状物。将该黄色油状物溶于10 ml 甲醇,加入3eq NaBH4,室温搅拌2h,0 oC下加入1 ml饱和NH4Cl溶液和30 ml乙酸乙酯搅拌分层,有机相用饱和NaCl溶液洗涤3次,无水Na2SO4干燥,减压蒸除溶剂,得到淡黄色油状物。该油状物0.5 g溶于10 ml 二氯甲烷,0 oC下加入3eq mCPBA,搅拌45 min,硅藻土过滤,加入60 ml乙酸乙酯,用5%NaOH溶液与饱和NaCl溶液各洗涤3次,有机相用无水Na2SO4干燥,减压蒸除溶剂,得到白色固体,该白色固体经硅胶柱层析(石油醚:乙酸乙酯=15:1~3:1)得到化合物11为白色固体。 ESI: 475 [M+23]+, 1H NMR (CDCl3, 600 MHz) 特征信号δ 4.76 (dd, J = 15.8, 6.8 Hz, 2H), 4.61 (t, J = 6.9 Hz, 2H), 4.10 – 4.06 (m, 1H), 3.82 – 3.76 (m, 1H), 3.39 (t, J = 3.3 Hz, 7H), 3.20 (d, J = 3.0 Hz, 1H), 3.06 (dd, J = 11.8, 4.5 Hz, 1H), 1.98 (dd, J = 9.9, 4.3 Hz, 1H), 1.84 (dd, J = 11.8, 8.6 Hz, 2H), 1.77 – 1.71 (m, 2H), 1.71 – 1.65 (m, 3H), 1.65 – 1.58 (m, 4H), 1.58 – 1.49 (m, 4H), 1.40 (ddd, J = 21.1, 17.4, 7.7 Hz, 3H), 1.20 (d, J = 9.6 Hz, 7H), 0.97 (s, 3H), 0.90 – 0.85 (m, 9H), 0.83 (t, J = 8.9 Hz, 9H)。
实施例7:
化合物12的制备:
将0.2 g 化合物11溶于5 ml四氢呋喃,-0 oC下加入1.2eq NaH,加完后室温下搅拌1h,滴加1.2eq MeI的四氢呋喃溶液2 ml,室温搅拌反应2h,加入2 ml饱和NH4Cl溶液与10 ml乙酸乙酯搅拌分层,有机相用饱和NaCl溶液洗涤3次,无水Na2SO4干燥,减压蒸除溶剂,得到淡黄色油状物。将该淡黄色油状物溶于5 ml 四氢呋喃,0 oC下加入2eq LiAlH4,在该温度下搅拌2h,0 oC下加入0.1 ml水和0.3 ml 15%NaOH溶液, 0.3 ml水,搅拌,过滤,滤饼用乙酸乙酯洗,有机相用饱和NaCl溶液洗涤3次,无水Na2SO4干燥,减压蒸除溶剂,得到白色固体,该白色固体经硅胶柱层析(石油醚:乙酸乙酯=10:1~4:1)得到化合物12为白色固体。 ESI: 491 [M+23]+, 1H NMR (CDCl3, 500 MHz) 特征信号δ 4.73 (dd, J = 15.8, 6.8 Hz, 2H), 4.61 (t, J = 6.9 Hz, 2H), 4.43 (s, 1H), 4.20 (s, 1H), 3.50 (s, 1H), 3.40 (d, J = 5.0 Hz, 9H), 3.20 (s, 1H), 2.05 – 1.06 (m, 15H), 0.86 (s, 3H)。
实施例8:
化合物3的制备:
将0.2 g 化合物12溶于5 ml DMSO,加入2eq IBX,室温下搅拌2h,加入2 ml饱和Na2S2O3溶液与10 ml乙酸乙酯搅拌分层,有机相用饱和NaCl溶液洗涤3次,无水Na2SO4干燥,减压蒸除溶剂,得到白色固体。将该白色固体溶于10 ml 二氯甲烷,0 oC下加入2eq TFA,在该温度下搅拌2 h,加入5 ml 15% NaOH溶液,搅拌分层,有机相用饱和NaCl溶液洗涤3次,无水Na2SO4干燥,减压蒸除溶剂,得到白色固体。该白色固体溶于4 ml二氯甲烷,加入1.2 eq TEA与1.1 eq 醋酐,搅拌反应8 h,加入饱和NaCl溶液,搅拌分层,有机相用饱和NaCl溶液洗涤3次,无水Na2SO4干燥,减压蒸除溶剂,得到白色固体,该白色固体经硅胶柱层析(石油醚:乙酸乙酯=8:1~2:1)得到化合物3为白色固体。 ESI: 443 [M+23]+
实施例9:
化合物4的制备:
将0.02 g 化合物3溶于1 ml 甲醇,加入2eq K2CO3,室温下搅拌6 h,加入2 ml饱和NaCl溶液与10 ml乙酸乙酯搅拌分层,有机相用饱和NaCl溶液洗涤3次,无水Na2SO4干燥,减压蒸除溶剂,得到白色固体,该白色固体经硅胶柱层析(石油醚:乙酸乙酯=5:1~1:1)得到化合物4为白色固体。 ESI: 401 [M+23]+
实施例10:
化合物6的制备:
将0.02 g 化合物3溶于1 ml 甲苯,加入3eq DBU,60 oC下搅拌4 h,加入2 ml饱和NaCl溶液与10 ml乙酸乙酯搅拌分层,有机相用饱和NaCl溶液洗涤3次,无水Na2SO4干燥,减压蒸除溶剂,得到黄色油状物,该黄色油状物经硅胶柱层析(石油醚:乙酸乙酯=12:1~4:1)得到化合物6为无色油状物。 ESI: 411 [M+23]+
实施例11:
化合物5的制备:
将0.02 g 化合物6溶于1 ml甲醇,加入2eq K2CO3,室温下搅拌6 h,加入2 ml饱和NaCl溶液与10 ml乙酸乙酯搅拌分层,有机相用饱和NaCl溶液洗涤3次,无水Na2SO4干燥,减压蒸除溶剂,得到淡黄色油状物,该油状物经硅胶柱层析(石油醚:乙酸乙酯=5:1~1:1)得到化合物5为无色油状物。 ESI: 369 [M+23]+
实施例12:
化合物15的制备:
将1 g 化合物3溶于20 ml 四氢呋喃与20 ml叔丁醇,0 oC下缓慢加入0.5 g NaBH4,加完后缓慢升温至40 oC搅拌10h,0 oC下加入5 ml饱和NH4Cl溶液,乙酸乙酯稀释,用饱和NaCl溶液洗涤3次,有机相用无水NaSO4干燥,减压蒸除溶剂,得到白色固体。
上述的白色固体0.6 g 溶于20 ml甲苯,升温至90 oC,加入Burgess’Reagent 0.34 g,该溶液回流3小时,冷却至室温,乙酸乙酯稀释,饱和NaCl溶液洗涤3次,有机相用无水NaSO4干燥,减压蒸除溶剂,得到白色固体。
上述的白色固体0.4 g溶于3 ml CCl4,加入5 ml饱和NaHCO3溶液与0.86 g NaIO4,搅拌15 分钟, 加入0.021 g RuCl3·nH2O的MeCN溶液3 ml,上述混合物在室温下避光搅拌7天, 硅藻土过滤,加入20 ml乙酸乙酯,用饱和NaCl溶液洗涤3次,有机相用无水Na2SO4干燥,减压蒸除溶剂,得到黄色油状物,该油状物经硅胶柱层析(石油醚:乙酸乙酯=10:1)得到白色针状晶体化合物15。
ESI: 455 [M+23]+, 1H-NMR( CDCl3,500MHz) 特征信号 δ:4.98-5.04(1H, td, J=5.4/10.8/10.8, H-C(12)),4.47-4.50(1H, dd, J=4.6/11.6, H-C(3)),2.36(1H, d, J=10.8, H-C(13)),2.10-2.30(2H, m, H-C(16))。
实施例13:
化合物16的制备:
将0.1 g 化合物V′溶于5 ml 二氯甲烷,0 oC下加入0.181 ml三乙胺,搅拌10分钟,缓慢滴加TMSOTf 0.118ml,加完后在室温反应4小时,乙酸乙酯稀释,用饱和NaCl溶液洗涤3次,有机相用无水NaSO4干燥,减压蒸除溶剂,得到淡黄色固体。
上述淡黄色固体0.14 g溶于4 ml二氯甲烷,将该溶液在0 oC下缓慢加入0.894 g KHSO4与0.141 g 间氯过氧苯甲酸的2 ml二氯甲烷溶液中,室温反应8小时,乙酸乙酯稀释,用饱和Na2S2O3溶液洗涤3次,饱和NaCl溶液洗涤3次,有机相用无水NaSO4干燥,减压蒸除溶剂,得到淡黄色油状物。
上述淡黄色油状物0.07 g溶于4 ml四氢呋喃,0 oC下缓慢滴加0.16 ml 1mol/L四丁基氟化铵的四氢呋喃溶液,室温反应2小时,乙酸乙酯稀释,用饱和NaCl溶液洗涤3次,有机相用无水NaSO4干燥,减压蒸除溶剂,得到淡黄色固体。该固体经硅胶柱层析(石油醚:乙酸乙酯=4:1~2:1)得到白色针状晶体化合物16。ESI: 498 [M+23]+, 1H-NMR( CDCl3,500MHz) 特征信号 δ:4.71-4.78(1H, td, J=5.2/11 /11, H-C(12)),4.45-4.49(1H, dd, J=4.6/11.5, H-C(3)),4.12-4.26(1H, qd, J=4.6/19.1 /19.1/19.1, H-C(21)),3.14-3.17(1H, t, J=4.6, C(21)-OH),2.78-2.85(1H, td, J=6.2/10.7/10.7, H-C(17)),2.39-2.44(1H, t, J=11.1, H-C(13))。
实施例14:
化合物17的制备:
将2 g化合物13溶解于50 ml THF中,0 oC下加入2 eq NaH搅拌30 min,2eq的BnBr加入上述溶液中,60 oC反应过夜,0 oC下乙酸乙酯稀释,缓慢滴加5 ml水,饱和NaCl溶液洗3次,有机层蒸干得淡黄色蜡状物。该蜡状物经硅胶柱层析(石油醚:乙酸乙酯=20:1~10:1)得到化合物17为白色固体。
实施例15:
化合物18的制备:
将2 g化合物13溶解于50 ml DCM中,0 oC下加入7.2eq DIPEA搅拌30 min,7.2eq的MOMOCl加入上述溶液中,室温反应1 h,饱和NaCl溶液洗3次,有机层蒸干得黄色油状物。该黄色油状物经硅胶柱层析(石油醚:乙酸乙酯=40:1~20:1)得到化合物18为淡黄色油状物。ESI: 573 [M+23]+, 1H NMR (500 MHz, CDCl3) 特征信号δ 4.82 (dd, J = 10.3, 7.3 Hz, 1H), 4.71 (dd, J = 15.3, 7.1 Hz, 1H), 4.59 (t, J = 6.5 Hz, 2H), 3.55 (dt, J = 18.5, 8.6 Hz, 1H), 3.42 – 3.31 (m, 7H), 3.06 (dd, J = 11.7, 4.1 Hz, 1H), 2.06 – 1.96 (m, 2H), 1.93 – 1.83 (m, 2H), 1.71 (dd, J = 19.1, 11.3 Hz, 3H), 1.60 (d, J = 8.9 Hz, 1H), 0.90 – 0.81 (m, 12H), 0.79 (s, 3H).
实施例16:
化合物19的制备:
将0.1 g化合物15溶解于4 ml toluene中,室温下加入2eq IBX与0.01eq PTSA搅拌10 min,接着80 oC反应8 h,乙酸乙酯稀释,饱和NaCl溶液洗3次,有机层蒸干得黄色油状物。该黄色油状物经硅胶柱层析(石油醚:乙酸乙酯=10:1~6:1)得到化合物19为淡黄色油状物。ESI: 411 [M+23]+, 1H NMR (600 MHz, CDCl3) 特征信号 δ 7.45 (dd, J = 12.4, 5.9 Hz, 1H), 6.15 (d, J = 6.0 Hz, 1H), 4.52 (dd, J = 11.4, 4.6 Hz, 1H), 3.62 (d, J = 4.8 Hz, 1H), 2.36 – 2.26 (m, 1H), 2.22 – 2.13 (m, 2H).
实施例17:
化合物20的制备:
将0.1 g化合物15溶解于4 ml THF/MeOH(3.8/0.2)中,室温下加入2eq NaBH4搅拌反应6 h,乙酸乙酯稀释,饱和NaCl溶液洗3次,有机层蒸干得白色固体。该白色固体经硅胶柱层析(石油醚:乙酸乙酯=10:1~3:1)得到化合物20为白色固体。ESI: 415 [M+23]+, 1H NMR (400 MHz, CDCl3) 特征信号δ 4.48 (dd, J = 10.9, 5.8 Hz, 2H), 3.91 (td, J = 10.7, 4.8 Hz, 1H), 2.29 – 2.09 (m, 3H), 2.05 (s, 4H), 1.88 (d, J = 10.7 Hz, 1H), 1.68 (dt, J = 16.9, 11.9 Hz, 5H), 1.62 – 1.38 (m, 8H), 1.27 (d, J = 15.5 Hz, 9H), 1.10 (s, 5H), 0.88 (dd, J = 31.1, 12.8 Hz, 17H).
实施例18:
化合物21的制备:
将0.1 g化合物16溶解于4 ml DCM中,-78 oC下加入2.5eq DAST搅拌反应1.5 h,1 ml饱和NH4Cl溶液加入,乙酸乙酯稀释,饱和NaCl溶液洗3次,有机层蒸干得黄色固体。该黄色固体经硅胶柱层析(石油醚:乙酸乙酯=20:1~15:1)得到化合物21为白色固体。ESI: 501 [M+23]+, 1H NMR (400 MHz, CDCl3) 特征信号δ 4.86 (s, 1H), 4.81 – 4.69 (m, 2H), 4.48 (dd, J = 11.4, 4.3 Hz, 1H), 3.04 (d, J = 6.2 Hz, 1H), 2.40 (dd, J = 22.1, 11.0 Hz, 1H), 2.06 (d, J = 13.3 Hz, 4H), 1.96 – 1.85 (m, 4H), 1.79 (dd, J = 21.1, 10.3 Hz, 1H), 1.65 (dd, J = 16.1, 7.6 Hz, 3H), 1.62 – 1.42 (m, 7H), 1.38 – 1.16 (m, 7H), 1.09 (d, J = 16.8 Hz, 4H), 0.94 (d, J = 15.0 Hz, 3H), 0.90 (s, 3H), 0.84 (d, J = 10.0 Hz, 7H).
实施例19:
化合物22的制备:
将0.1 g化合物15溶解于4 ml THF/MeOH(3.8/0.2)中,室温下加入2eq NaBH4搅拌反应6 h,乙酸乙酯稀释,饱和NaCl溶液洗3次,有机层蒸干得白色固体。该白色固体溶于4 ml 吡啶中,0 oC下加入4eq POCl3搅拌10 min,接着40 oC反应6 h,乙酸乙酯稀释,饱和NaCl溶液洗3次,有机层蒸干得黄色油状物。该黄色油状物经硅胶柱层析(石油醚:乙酸乙酯=30:1~20:1)得到化合物22为无色油状物。ESI: 439 [M+23]+, 1H NMR (400 MHz, CDCl3) 特征信号δ 5.37 (s, 1H), 5.26 (s, 1H), 4.50 (dd, J = 11.1, 5.1 Hz, 1H), 2.32 (d, J = 24.1 Hz, 1H), 2.25 – 2.15 (m, 1H), 2.08 (d, J = 20.9 Hz, 5H), 1.96 – 1.82 (m, 1H), 1.77 – 1.46 (m, 8H), 1.46 – 1.33 (m, 6H).
实施例20:
化合物23的制备:
将0.1 g化合物15溶解于4 ml THF/MeOH(3.8/0.2)中,室温下加入2eq NaBH4搅拌反应6 h,乙酸乙酯稀释,饱和NaCl溶液洗3次,有机层蒸干得白色固体。该白色固体溶于4 ml 吡啶中,0 oC下加入4eq POCl3搅拌10 min,接着40 oC反应6 h,乙酸乙酯稀释,饱和NaCl溶液洗3次,有机层蒸干得黄色油状物。该黄色油状物经硅胶柱层析(石油醚:乙酸乙酯=30:1~20:1)得到化合物23为无色油状物。ESI: 476 [M+23]+, 1H NMR (600 MHz, CDCl3) 特征信号δ 5.07 (td, J = 10.7, 5.1 Hz, 1H), 4.53 – 4.44 (m, 1H), 4.19 (td, J = 9.6, 4.3 Hz, 1H), 2.53 – 2.44 (m, 1H), 2.26 – 2.20 (m, 1H), 2.18 (s, 2H), 2.11 – 2.07 (m, 1H), 2.05 (dd, J = 8.4, 2.1 Hz, 5H), 2.02 (dd, J = 10.5, 4.0 Hz, 2H), 1.92 – 1.85 (m, 2H), 1.81 – 1.73 (m, 1H).
实施例21:
化合物24的制备:
将1 g化合物3溶解于10 ml THF/MeOH(9.9/0.1)中,0 oC下加入3eq NaBH4搅拌30 min,接着室温反应2 h,0 oC下加入乙酸乙酯稀释与2 ml饱和NH4Cl溶液,饱和NaCl溶液洗3次,有机层蒸干得白色固体。该白色固体经硅胶柱层析(石油醚:乙酸乙酯=12:1~8:1)得到化合物24为白色固体。ESI: 485 [M+23]+, 1H NMR (600 MHz, CDCl3) 特征信号δ 4.86 (td, J = 10.9, 5.1 Hz, 1H), 4.48 (dd, J = 11.4, 4.7 Hz, 1H), 4.39 – 4.33 (m, 1H), 2.15 – 2.09 (m, 1H), 2.08 – 2.01 (m, 6H), 1.92 – 1.87 (m, 1H), 1.75 (ddd, J = 11.5, 6.9, 3.6 Hz, 2H), 1.70 – 1.58 (m, 5H), 1.57 – 1.52 (m, 1H), 1.51 – 1.41 (m, 6H), 1.35 – 1.29 (m, 1H), 1.26 (t, J = 11.8 Hz, 2H), 1.14 – 1.06 (m, 2H), 1.03 (s, 3H), 0.94 – 0.81 (m, 13H).
实施例22:
化合物25的制备:
将0.5 g化合物24溶解于5 ml 吡啶中,0 oC下加入4eq POCl3搅拌10 min,接着40 oC反应6h,乙酸乙酯稀释,饱和NaCl溶液洗3次,有机层蒸干得黄色油状物。该黄色油状物经硅胶柱层析(石油醚:乙酸乙酯=20:1~12:1)得到化合物25为无色油状物。ESI: 504[M+23]+, 1H NMR (600 MHz, CDCl3) 特征信号δ 4.86 (td, J = 10.9, 5.1 Hz, 1H), 4.48 (dd, J = 11.4, 4.7 Hz, 1H), 4.39 – 4.31 (m, 1H), 2.16 – 2.08 (m, 1H), 2.08 – 2.01 (m, 6H), 1.90 (ddd, J = 12.3, 10.4, 7.1 Hz, 1H), 1.79 – 1.71 (m, 2H), 1.71 – 1.58 (m, 5H), 1.58 – 1.51 (m, 1H), 1.51 – 1.41 (m, 6H), 1.35 – 1.20 (m, 3H), 1.15 – 1.07 (m, 2H), 1.03 (s, 3H), 0.89 (dd, J = 15.2, 9.3 Hz, 6H), 0.87 – 0.80 (m, 7H).
实施例23:
化合物26的制备:
将1 g化合物3溶解于10 ml MeOH中,加入3eq K2CO3 40 oC下搅拌4 h,蒸干溶剂得淡黄色固体。该淡黄色固体经硅胶柱层析(石油醚:乙酸乙酯=6:1~2:1)得到化合物26为白色固体。ESI: 441 [M+23]+, 1H NMR (400 MHz, CDCl3) 特征信号δ 4.47 (dd, J = 10.6, 5.8 Hz, 1H), 3.47 (d, J = 4.4 Hz, 1H), 2.86 (td, J = 11.0, 5.5 Hz, 1H), 2.57 (d, J = 5.3 Hz, 1H), 2.21 (d, J = 14.1 Hz, 4H), 2.15 (dd, J = 12.3, 6.9 Hz, 1H), 2.04 (s, 3H), 1.87 (d, J = 8.1 Hz, 1H), 1.00 (d, J = 15.6 Hz, 4H), 0.94 – 0.79 (m, 15H).
实施例24:
化合物27的制备:
将1 g化合物3溶解于10 ml MeOH中,加入3eq K2CO3 50 oC下搅拌4 h,蒸干溶剂得淡黄色固体。该淡黄色固体经硅胶柱层析(石油醚:乙酸乙酯=6:1~2:1)得到化合物27为白色固体。ESI: 399 [M+23]+, 1H NMR (400 MHz, CDCl3) 特征信号δ 3.47 (s, 1H), 3.19 (dd, J = 11.2, 4.8 Hz, 1H), 2.85 (td, J = 11.0, 5.6 Hz, 1H), 2.60 (s, 1H), 2.20 (d, J = 14.7 Hz, 3H), 2.05 (t, J = 10.8 Hz, 1H), 1.88 (dd, J = 8.0, 3.1 Hz, 1H), 1.76 – 1.13 (m, 15H), 1.00 (d, J = 10.6 Hz, 3H), 0.97 (s, 4H), 0.88 (d, J = 3.5 Hz, 6H), 0.77 (s, 3H).
实施例25:
化合物28的制备:
将0.1 g化合物26溶解于3 ml DCM中,加入1.5eq PCC 室温下搅拌2 h,过滤,蒸干溶剂得黄色固体。该黄色固体经硅胶柱层析(石油醚:乙酸乙酯=15:1~10:1)得到化合物28为白色固体。ESI: 439 [M+23]+, 1H NMR (400 MHz, CDCl3) δ 4.47 (dd, J = 11.2, 5.2 Hz, 1H), 3.23 (td, J = 10.7, 6.7 Hz, 1H), 3.07 (d, J = 10.3 Hz, 1H), 2.28 – 2.16 (m, 6H), 2.10 – 1.99 (m, 5H), 1.89 – 1.76 (m, 2H), 1.76 – 1.48 (m, 11H), 1.41 (dd, J = 10.9, 7.6 Hz, 2H), 1.24 – 1.14 (m, 5H), 1.04 (ddd, J = 17.5, 11.3, 5.7 Hz, 2H), 0.98 – 0.92 (m, 4H), 0.90 – 0.82 (m, 9H), 0.79 (s, 3H).
实施例26:
化合物29的制备:
将0.1 g化合物26溶解于3 ml 吡啶中,0 oC下加入4eq POCl3搅拌10 min,40 oC反应6 h,乙酸乙酯稀释,饱和NaCl溶液洗3次,有机层蒸干得黄色油状物。该黄色油状物经硅胶柱层析(石油醚:乙酸乙酯=30:1~15:1)得到化合物29为无色油状物。ESI: 423 [M+23]+, 1H NMR (400 MHz, CDCl3) 特征信号δ 5.24 (d, J = 2.3 Hz, 1H), 4.50 (dd, J = 9.8, 6.3 Hz, 1H), 3.52 (s, 1H), 2.09 (d, J = 10.7 Hz, 3H), 2.06 (d, J = 10.6 Hz, 3H), 2.02 – 1.77 (m, 5H), 1.71 – 1.20 (m, 13H), 1.16 – 1.06 (m, 2H), 1.02 (d, J = 10.4 Hz, 3H), 0.98 (d, J = 8.5 Hz, 3H), 0.95 – 0.79 (m, 12H).
实施例27:
化合物30的制备:
将0.1 g化合物27溶解于3 ml DCM中,加入3eq PCC 室温下搅拌3 h,过滤,蒸干溶剂得黄色固体。该黄色固体经硅胶柱层析(石油醚:乙酸乙酯=15:1~10:1)得到化合物30为淡黄色固体。ESI: 395 [M+23]+, 1H NMR (400 MHz, CDCl3) 特征信号δ 3.31 – 3.17 (m, 1H), 3.12 (t, J = 10.0 Hz, 1H), 2.59 – 2.46 (m, 1H), 2.41 (ddd, J = 15.5, 7.1, 3.8 Hz, 1H), 2.23 (s, 3H), 2.09 (dt, J = 21.2, 8.1 Hz, 1H), 1.90 – 1.50 (m, 9H), 1.50 – 1.33 (m, 4H), 1.33 – 1.16 (m, 9H), 1.13 – 0.99 (m, 10H), 0.93 – 0.85 (m, 2H), 0.85 – 0.72 (m, 4H).
实施例28:
化合物31的制备:
将0.1 g化合物27溶解于3 ml 吡啶中,0 oC下加入8eq POCl3搅拌10 min,40 oC反应6 h,乙酸乙酯稀释,饱和NaCl溶液洗3次,有机层蒸干得黄色油状物。该黄色油状物经硅胶柱层析(石油醚:乙酸乙酯=30:1~15:1)得到化合物31为淡黄色油状物。ESI: 363 [M+23]+, 1H NMR (400 MHz, CDCl3) 特征信号δ 5.48 – 5.33 (m, 2H), 5.27 (d, J = 2.4 Hz, 1H), 3.52 (s, 1H), 2.84 – 2.63 (m, 1H), 2.09 (d, J = 11.5 Hz, 3H), 2.05 – 1.79 (m, 8H), 1.71 – 1.44 (m, 11H), 1.44 – 1.08 (m, 11H), 1.08 – 0.82 (m, 21H).
实施例29:
片剂的制备:
按本发明实施例2-28的方法先制得化合物1-31或无机酸(盐酸,硫酸,磷酸等)制成的盐,按其与赋形剂重量比为1:5-1:10的比例加入赋形剂,制粒压片即可。
达玛烷三萜衍生物及其药物组合物和其在制药中的应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。













动态评分
0.0