









专利摘要
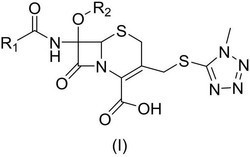
本发明涉及头孢烯衍生物及其制备方法,它公开了一种具有抗微生物活性的头孢酯脒,即化学式I所表征的头孢烯化合物及其药物上可接受的盐。该头孢烯化合物是将头孢硫脒溶解在适当的溶剂中,用酸催化水解和缩合,反应一步完成。该化合物及其盐性质稳定,纯度高且具有较好的抗菌活性。本发明还提供以其为活性成分的药物组合物及在人类和动物中治疗疾病感染中的应用。
权利要求
1、结构式I所示的头孢烯衍生物及其生理上或是药理上相容的盐类。
2、权利要求1所述的头孢烯衍生物,其特征为无机酸加成盐或有机酸加成盐类。
3、权利要求2所述的头孢烯衍生物,其中无机酸加成盐类优选盐酸盐,硫酸盐,乳酸盐或柠檬酸盐。
4、权利要求1-3之一所述的头孢烯衍生物的制备方法,其特征是将头孢硫脒溶解在适当的溶剂中,用酸催化水解和缩合,使反应一步完成。
5、权利要求2-3之一所述的头孢烯衍生物的制备方法,其特征是通过将化合物I加入适合的酸得到化合物I的盐。
6、权利要求4所述的头孢烯衍生物的制备方法,其特征是反应的温度范围为0℃至40℃,反应时间为12小时至48小时。
7、权利要求4或5所述的头孢烯衍生物的制备方法,其特征是所述的酸为有机酸、无机酸或路易氏酸。
8、权利要求7所述的头孢烯衍生物的制备方法,其特征是:
有机酸为甲酸、乙酸、丙酸、三氯乙酸、三氟乙酸等;
无机酸为盐酸、溴氢酸、硫酸、氯化氢、溴化氢等;
路易氏酸为三氯乙酸、三氟乙酸等,用路易氏酸反应时使用阳离子捕集剂,阳离子捕集剂为苯甲醚、苯酚。
9、权利要求4所述的头孢烯衍生物的制备方法,其特征是所述的溶剂为水或与水互溶的有机溶剂包括酰胺类,酮类,腈类,醇类,或水与有机溶剂的混合物溶剂。
10、权利要求9所述的头孢烯衍生物的制备方法,其特征是所述的溶剂为甲醇、乙醇、异丙醇、丙酮、乙腈、DMF。
11、一种抗菌药物组合物,包括权利要求1-3之一所述的头孢烯衍生物作为活性成分与药学上可接受的赋形剂或载体。
12、权利要求11的抗菌药物组合物,其特征是口服制剂或注射制剂。
说明书
技术领域技术领域
本发明涉及具有抗微生物活性的头孢烯化合物及其药物上可接受的盐;具体的说是涉及具有硫脒乙酰侧链的头孢菌素及其药物上可接受的盐。
技术背景背景技术
β-内酰胺类抗生素(β-lactams)系指化学结构中具有β-内酰胺环的一大类抗生素,包括临床最常用的青霉素与头孢菌素,以及新发展的头霉素类、硫霉素类、单环β-内酰胺类等其他非典型β-内酰胺类抗生素。此类抗生素具有杀菌活性强、毒性低、适应症广及临床疗效好的优点。通过对本类化合物的化学结构修饰,即通过对β-内酰胺母核三、四及七位的修饰,得到了作用各有特点的多种β-内酰胺类抗生素,形成了许多不同抗菌谱和抗菌作用以及各种临床药理学特性的抗生素。
头孢硫脒是第一代头孢菌素,具有抗菌谱广,抗菌作用强(特别是对肠球菌、金黄色葡萄球菌),血药浓度高,组织分布广,临床疗效好,副作用少等特点。尽管在临床使用中,头孢硫脒是一种优良的第一代头孢烯抗生素,但在使用和运输贮存过程中,发现头孢硫脒性质不稳定,放置过程中,容易色泽加深和含量降低。
究其原因,首先是因为头孢硫脒的三位带有一个乙酰基,容易被水解破坏;另外,头孢硫脒在四位还带有一个相当活泼的羧基,容易受到外界各种因素的影响,引起头孢硫脒本身结构的变化,使头孢硫脒发生聚合或降解,导致产品的色泽加深、含量下降和杂质增多,影响到头孢硫脒的疗效。另外头孢硫脒本身是一个内盐,具备优良的水溶性,只适于制备成注射剂使用,在用药形式方面也较为单一。
我们针对头孢硫脒的不足进行了结构修饰,发现通过对头孢硫脒的β-内酰胺母核的三、四位进行修饰后,得到一种新的头孢烯类化合物I,化合物I与头孢硫脒相比,其结构上的显著特征是头孢烯母核上的三位和四位形成一个稳定的内酯环。这种内酯环结构相对头孢硫脒的三位和四位来说是非常稳定的。
这个新的头孢烯类化合物I及其盐和他们的合成方法未见公开。本发明成功地合成了化合物I,并且观察了它的抗菌活性和抗菌谱,发现化合物I对各种细菌具有较好的抗菌作用。
发明内容发明内容
本发明的一个目的是提供具有抗微生物高度活性的头孢烯化合物及其药物上可接受的盐。
本发明另一个目的是提供该头孢烯化合物及其药物上可接受的盐的制备方法。
本发明进一步的目的是提供一个药物组合,其中包括以该头孢烯化合物及其药物上可接受的盐作为活性成分。
本发明还有一个目的是提供该头孢烯化合物及其药物上可接受的盐在治疗由于病原微生物引起的疾病感染中的应用。
本发明的技术方案如下:
本发明提供一种头孢烯化合物,可以用下面的结构式表示:
化合物I(头孢酯脒)可用如下参数表征:
1HNMR(DMSO-d6,500Hz):
1.23(m,12H, 3.29(s,3H, 3.83,4.25(d,2H,J=13.0Hz,C2-H),5.05(s,2H, 5.14(d,1H,J=5.0Hz,C6-H),5.85(d,1H,J=4.5Hz,C7-H),9.40(d,1H,J=3.0Hz,
13CNMR(DMSO-d6,100Hz):
35.1(t,C-2),142.9(s,C-3),122.9(s,C-4),60.2(d,C-6),57.0(d,C-7),163.4(s,C-8),168.6(s, 71.7(t, 166.8(s, 22.7(t, 163.4(s, 49.7(d, 21.4(q, 46.6(d, 22.3(q,
IR(KBr,cm-1):3427、3293、3233、2977,1774,1746,1670,1652,1623,1589,1553,1419,1319,1147,1020,983,677,526。
ESIMS(m/e):413.3[M+H]+
FABMS(m/e):413[M+H]+
X-粉末衍射图谱表明为结晶态
X-衍射图谱数据:
2θ d I/I0
9.46 9.34 1.00
13.00 6.80 0.21
18.98 4.67 0.39
21.00 4.23 0.28
22.20 4.00 0.37
溶解性:本化合物在水、甲醇中略溶,在无水乙醇、丙酮、二氯甲烷及乙醚中不溶。
化合物I结构上带有碱性基团,通过和酸成盐后又可得到水溶性的化合物I的盐,这样化合物I的盐可以制备成注射剂用于治疗感染。
因此,本发明化合物的盐的制备方法为将化合物I加入适合的酸而得到。
化合物I可以是生理上或是药理上相容的盐类,如无机酸加成盐和有机酸加成盐类。以上化合物I的盐类均包括在本发明的范围内。
化合物的无机酸加成盐类如盐酸盐,氢溴酸盐,硫酸盐,乳酸盐、柠檬酸盐。硝酸盐或磷酸盐;优选盐酸盐、硫酸盐、乳酸盐或柠檬酸盐。
化合物I的盐,以化合物I的硫酸盐为例,X-粉末衍射图谱表明其为结晶态。其X-衍射图谱数据如下:
2θ d I/I0
7.78 11.35 0.38
9.26 9.54 1.00
13.42 6.59 0.17
15.62 5.67 0.19
17.98 4.9 0.21
18.60 4.77 0.67
21.82 4.07 0.26
23.52 3.78 0.35
有机酸加成盐如对甲苯磺酸盐,甲磺酸盐,甲酸盐,三氟乙酸盐或马来酸盐。
由于化合物I的β-内酰胺环的碳三位和四位形成了一个稳定的内酯环,在结构上没有性质活泼的羧基和乙酰基暴露在外,所以化合物I对酸碱稳定,特别是在强酸性的条件下表现出比头孢硫脒更加稳定的特性。我们知道,人体的胃液是强酸性的,通过结构修饰后得到的化合物I在人体的胃液中是稳定的。
另外化合物I相对头孢硫脒而言,头孢硫脒为水溶性的,而化合物I是水不溶性的,且对强酸极为稳定,这表明化合物I可以用于口服治疗由于病原微生物引起的疾病感染;
本发明头孢烯衍生物的制备方法,是将头孢硫脒式II溶解在适当的溶剂中,用酸催化水解和缩合,使反应一步完成。
在制备过程中,主要是通过水解化合物II三位的酰基得到羟甲基,羟甲基再与四位的羧基缩合得到内酯环。这个过程并不是轻易得到的。由于两步反应是在一个体系中完成的,水解和酯化过程极为缓慢,通过实验摸索和调节实验方案,我们成功地合成出纯度高质量好的化合物I。由于头孢硫脒在强酸强碱条件下极不稳定,特别是较高温度(大于20度)下头孢硫脒本身的母核结构容易被破坏,导致反应液色泽变深,产生大量的杂质;但如果反应在太低的温度下进行,我们所需要的水解和酯化过程不容易得到。通过实验,发现可以在较高温度下,通过反应避光进行,则反应物易于完成,且原料在反应过程中不易降解和聚合,产品质量好,杂质少,得率高。
上述反应时间为1小时至72小时,优选为12至48小时。
上述反应温度为-20℃至60℃,优选为0-40℃。
本发明所用的酸包括有机酸,优选甲酸、乙酸、丙酸、三氯乙酸、三氟乙酸等;
或无机酸,优选盐酸、溴氢酸、硫酸、氯化氢、溴化氢等;
或路易氏酸,优选三氯乙酸、三氟乙酸等。
本发明用路易氏酸进行反应时最好在阳离子捕集剂存在下进行,所用的阳离子捕集剂优为苯甲醚、苯酚。
本发明不排除用合适的碱作为催化剂,包括无机碱和有机碱:如碱金属(如钾、钠等)、碱土金属(如钙、镁等)的碳酸盐或碳酸氢盐、有机碱为三烷基胺如三甲胺,三乙胺,甲基吡啶等等。
本发明水解和缩合反应通常在常用的对反应无不良影响的溶剂存在或无溶剂存在下进行。对反应来说,任何不妨碍本反应的溶剂都可使用,如醚类,酯类,卤代烃类,烃类,酰胺类,酮类,腈类,醇类和水或它们之中任意的混合物。
所述的溶剂优选为水或与水互溶的有机溶剂包括酰胺类、酮类,腈类,醇类,或水与有机溶剂的混合物溶剂。
上述的溶剂为甲醇、乙醇、异丙醇、丙酮、乙腈、DMF。
此外,本发明中用作反应的酸、碱如果为液体时,其本身也能用作溶剂。
为了说明化合物1的用途,本发明的化合物的MIC(最低抑菌浓度)的实验数据在下面表示。
试验方法:采取常规的体外抑菌试验方法
试验数据:
细菌名称 最低抑菌浓度MIC(mg/L)
金葡萄球菌 ≤2
表皮葡萄球菌 ≤2
肠球菌 16-128
肺炎链球菌 0.25-8
流感嗜血杆菌 4-256
溶血性链球菌 ≤2
沙门菌属 8-64
通过以上实据,可以知道化合物I对革兰氏阳性菌有较好的抑菌活性,对耐药肠球菌及沙门菌属有一定抑菌活性。
本发明还提供一种抗菌药物组合物,包括所述的头孢烯衍生物作为活性成分与药学上可接受的赋形剂或载体。
上述抗菌药物组合物,其特征是口服制剂或注射制剂。
本发明化合物I,可以是口服的如胶囊,片剂或颗粒剂;或非口服如注射剂,像已知的青霉素和头孢菌素制剂一样施用。注射剂的载体可举例如蒸馏水或生理盐水。当作为胶囊、粉剂、颗粒剂或片剂使用时,化合物I与常规的药理上可相容的赋形剂(例如,淀粉,麦芽糖,蔗糖,碳酸钙或磷酸钙),粘合剂(例如淀粉,阿拉伯胶,羧甲基纤维素,羟丙基纤维素或结晶纤维素),润滑剂(例如硬脂酸镁或滑石粉)和分解剂(例如,羧甲基钙或滑石粉)相混合。
含有化合物I的药物组合物是用已知方法制备的。该组合物通常是这样制取的,即将至少一种化合物I或它的盐与上述的载体或赋形剂相混合。化合物I占整个组合物的比率一般为5-10%(W/W),对固体组合物如胶囊,片剂和颗粒剂以20-100%(W/W)为较好,对液体组合物如注射剂等,以5-30%(W/W)为较好。
本发明的有益效果在于化合物I从抗菌活性来说,其具有较宽的抗革兰氏阳性菌的活性;从其理化性质来说,化合物I具有脂溶性,可以考虑口服给药,降低了治疗成本,用药方便;另外化合物I的盐具有水溶性,可以考虑制备成注射剂,体现出注射剂起效快的特点。这样从用药途径上来说,与头孢硫脒相比,头孢硫脒只能用于注射,用药途径单一,而化合物I的用药途径更为广阔,所以化合物I及其盐在抗感染治疗的领域中具备独特的治疗特点。
另外化合物I性质稳定,纯度高,适于贮藏和运输,而不容易引起色泽加深、含量降低,这从另一方面保证了化合物I实际使用过程中保存了其优良的抗菌性。
附图说明具体实施方式下面是实施例,本发明的实施例并不局限于这些实施例。
化合物I(头孢酯脒)的制备
实施例一
30-40℃下,反应器中加入5克头孢硫脒,加入8ml水及20ml乙腈溶解,过滤,滤液中加入2.5ml12N盐酸,保温搅拌12小时后,用5%氢氧化钠水溶液调节溶液酸度在2-4之间,搅拌3小时后过滤,,乙腈洗涤两次,抽干,减压干燥,得到化合物I3.15克,为白色或类白色结晶,220℃以上分解,纯度(HPLC归一法)为98.1%。
1HNMR(DMSO-d6,500Hz):
1.23(m,12H, 3.29(s,3H, 3.83,4.25(d,2H,J=13.0Hz,C2-H),5.05(s,2H, 5.14(d,1H,J=5.0Hz,C6-H),5.85(d,1H,J=4.5Hz,C7-H),9.40(d,1H,J=3.0Hz,
IR(KBr,cm-1):
3427,3293,3233,2977,1774,1746,1670,1652,1623,1589,1553,1419,1319,1147,1020,983,677,526。
X-衍射图谱数据:
2θ d I/I0
9.46 9.34 1.00
13.00 6.80 0.21
18.98 4.67 0.39
21.00 4.23 0.28
22.20 4.00 0.37
实施例二
10-20℃下,反应器中加入5克头孢硫脒,加入10ml水及15ml甲醇溶解,过滤,滤液中加入3ml12N盐酸,搅拌30小时后,用10%氨水溶液调节溶液酸度在2-4之间,搅拌4小时后过滤,乙醇洗涤两次,抽干,减压干燥,得到化合物I3.05克。
化合物与例一的化合物的IR(KBr,cm-1)及1HNMR(DMSO-d6,500Hz)相同
实施例三
常温下,反应器中加入5克头孢硫脒,加入8ml水及20ml丙酮及5ml乙酸乙酯溶解,过滤,滤液中加入5ml磷酸,35-40℃搅拌20小时后,用5%氢氧化钠水溶液调节溶液酸度在2-4之间,搅拌3小时后过滤,丙酮洗涤两次,抽干,减压干燥,得到化合物I2.8克。
实施例四
常温下,反应器中加入5克头孢硫脒,加入10ml水及15ml异丙醇醇溶解,过滤,滤液中加入4ml30%硫酸,避光静置48小时后,用8%氨水溶液调节溶液酸度至2-4,搅拌4小时后过滤,异丙醇洗涤两次,抽干,减压干燥,得到化合物I3.1克。
实施例五
0-10℃下,反应器中加入5克头孢硫脒,加入8ml水及20ml甲醇溶解,加入20ml甲酸,搅拌3小时后,避光室温放置72小时,然后用8%氨水调节溶液酸度在2-4之间,搅拌3小时后过滤,乙醇洗涤两次,抽干,减压干燥,得到化合物I2.7克。
实施例六
常温下,反应器中加入5克头孢硫脒,加入7ml水及20ml丙酮溶解,加入4ml三氟醋酸和1ml苯甲醚,搅拌10分钟后,避光15-20℃放置12小时,用5%氢氧化钠水溶液调节溶液酸度在2-4之间,搅拌2小时后过滤,丙酮洗涤两次,抽干,减压干燥,得到化合物I2.95克。
实施例七
5-10℃,反应器中加入5克头孢硫脒,加入10ml水及20ml乙醇溶解,加入3ml12N盐酸,升温至室温静置48小时后,用5%氢氧化钠水溶液调节溶液酸度在2-4之间,搅拌2小时后过滤,无水乙醇洗涤两次,抽干,减压干燥,得到化合物I3.1克。
实施例八
常温下,反应器中加入5克头孢硫脒,加入10ml水及20ml乙醇溶解,过滤,滤液中加入3ml12N盐酸,避光静置24小时后,用5%氢氧化钠水溶液调节溶液酸度至2-4,搅拌3小时后过滤,无水乙醇洗涤两次,抽干,减压干燥,得到化合物I2.95克,
实施例九
25-30℃下,反应器中加入5克头孢硫脒,加入10ml水及15ml乙醇溶解,过滤,滤液中加入5ml苯甲醚和5ml三氟乙酸,搅拌5小时,用5%氢氧化钠水溶液调节溶液酸度在2-4之间,搅拌2小时后过滤,无水乙醇洗涤两次,抽干,减压干燥,得到化合物I2.4克。
化合物I的盐的制备
实施例十
例一得到化合物I5克在常温下,加入反应器中,再加入6ml水及12ml醇搅拌悬浮,溶液中滴加20%的硫酸溶解固体,过滤,滤液中滴加无水乙醇至溶液浑浊,搅拌养晶30分钟后,再加入无水乙醇,80ml,常温养晶1小时,过滤,无水乙醇洗涤两次,抽干,减压干燥,得到化合物I硫酸盐4.3克,为白色或类白色结晶,纯度(HPLC归一法)为96.9%,206℃以上分解。
X-衍射图谱数据
2θ d I/I0
7.78 11.35 0.38
9.26 9.54 1.00
13.42 6.59 0.17
15.62 5.67 0.19
17.98 4.9 0.21
18.60 4.77 0.67
21.82 4.07 0.26
23.52 3.78 0.35
实施例十一
例一得到化合物I5克在常温下,加入反应器中,再加入8ml水及10ml丙酮搅拌悬浮,溶液中滴加3N盐酸溶解固体,过滤,滤液中滴加丙酮至溶液浑浊,搅拌养晶30分钟后,再加入丙酮50ml,常温养晶1小时,过滤,无水乙醇洗涤两次,抽干,减压干燥,得到化合物I盐酸盐4.1克。
实施例十二
例一得到化合物I5克在0-5℃下,加入反应器中,再加入10ml水及10ml异丙醇搅拌悬浮,溶液中滴加20%的硫酸溶解固体,过滤,滤液中滴加异丙醇至溶液浑浊,搅拌养晶1小时后,再加入异丙醇80ml,保温养晶2小时,过滤,异丙醇洗涤两次,抽干,减压干燥,得到化合物I硫酸盐4.2克。
实施例十三
例一得到化合物I5克在35-45℃下,加入反应器中,再加入30ml水搅拌悬浮,溶液中加入2.6克柠檬酸,搅拌溶解固体,过滤,滤液中滴加丙酮至溶液浑浊,搅拌养晶30分钟后,再加入丙酮300ml,降温到10-15℃养晶3小时,过滤,丙酮洗涤两次,抽干,减压干燥,得到化合物I柠檬酸盐3.1克。
实施例十四
常温下,反应器中加入例一得到化合物I5克和乳酸2克,,再加入35ml水,搅拌反应2小时,固体溶解,过滤,用5ml水洗涤反应器和过滤器,合并滤液,室温下溶液中滴加异丙醇至溶液浑浊,养晶2小时后,再滴加异丙醇200ml,降温5-10℃到养晶5小时,过滤,异丙醇洗涤两次,抽干,减压干燥,得到化合物I乳酸盐2.7克。
实施例十五
常温下,反应器中加入5克头孢硫脒,加入10ml水及15ml乙醇溶解,过滤,滤液中加入5ml12N盐酸,避光静置30小时后,降温至0-5℃,滴加无水乙醇150ml,析出大量固体,搅拌3小时后过滤,无水乙醇洗涤三次,抽干,减压干燥,得到化合物I的盐酸盐3.8克。
实施例十六
常温下,反应器中加入5克头孢硫脒,加入10ml水及15ml一,丙酮溶解,过滤,滤液中加入8ml30%盐酸,避光静置24小时后,降温至0-5℃,滴加丙酮100ml,析出大量固体,搅拌4小时后过滤,丙酮洗涤两次,抽干,减压干燥,得到化合物I的硫酸盐3.5克。
头孢烯衍生物及其制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。





动态评分
0.0