

IPC分类号 : B01J31/02,C07D229/00,C07D251/34,C07F9/6584





专利摘要
本发明涉及一种异氰酸酯聚合催化剂及其制备方法,及其用于制备聚异氰酸酯的方法。所述催化剂具有1,3,2‑二氮磷杂环戊烷结构,制备方法简单易行。将所述催化剂用于制备聚异氰酸酯,催化剂引发温度低,反应过程温和,单体转化率高,制备的聚异氰酸酯产品中兼有三聚体和二聚体结构。
权利要求
1.一种异氰酸酯聚合催化剂,其特征在于,所述催化剂的结构式为:
其中R1、R2、R3、R4相互独立的选自氢、苯基、具有1-10个碳原子的烷基、具有7-15个碳原子的烷苯基,优选氢、苯基、甲基、乙基、丙基、丁基、甲苯基、乙苯基;R5选自苯基、1-6个碳原子的烷基、1-6个碳原子的烷氧基、1-4个碳原子的烷氨基,优选苯基、甲基、乙基、正丙基、异丙基、甲氧基、乙氧基、二甲氨基、二乙氨基;
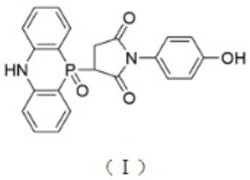
更优选的,所述催化剂选自结构式为式I、II、III、IV、V、VI的化合物中的一种或多种:
2.一种制备权利要求1所述的催化剂的方法,包括以下步骤:在无水无氧条件下,将N,N-二取代-1,2-二取代乙二胺 与一取代二氯化膦 在100-150℃加热混合,在160-190℃下反应1-3h。
3.根据权利要求2所述的方法,其特征在于,所述N,N-二取代-1,2-二取代乙二胺与一取代二氯化膦的摩尔比为1:1~1:1.5。
4.一种使用权利要求1所述的催化剂或权利要求2或3所述的方法制备的催化剂制备聚异氰酸酯的方法,包括以下步骤:在惰性气体的保护下,将异氰酸酯原料在所述催化剂的催化下进行自聚反应。
5.根据权利要求4所述的方法,其特征在于,所述自聚反应的温度为10-100℃,优选40-80℃。
6.根据权利要求4或5所述的方法,其特征在于,所述的催化剂的用量基于异氰酸酯原料的质量为100-1000ppm,优选300-600ppm。
7.根据权利要求4-6任一项所述的方法,其特征在于,所述催化剂的引发温度为40-50℃。
8.根据权利要求4-7任一项所述的方法,其特征在于,所述的异氰酸酯选自芳香族、脂肪族和脂环族二异氰酸酯中的一种或多种,优选苯二亚甲基二异氰酸酯、甲苯二异氰酸酯、丙烷二异氰酸酯、丁烷二异氰酸酯、戊基二异氰酸酯、六亚甲基二异氰酸酯、1,4-环己烷二异氰酸酯、异佛尔酮二异氰酸酯、1,4-双(异氰酸根合甲基)环己烷、2-甲基戊烷-1,5-二异氰酸酯、双(异氰酸根合甲基)降冰片烷和4,4’-二环己基甲烷二异氰酸酯中的一种或多种。
9.根据权利要求4-8任一项所述的方法,其特征在于,所述聚异氰酸酯产品的二聚体含量40-50wt%、三聚体含量45-60wt%、五聚体以上含量低于5wt%,基于五聚体以上多聚体、三聚体和二聚体的总重。
10.根据权利要求4-9任一项所述的方法,其特征在于,所述聚异氰酸酯产品的单体含量小于0.5wt%,基于100%固含无溶剂产品。
说明书
技术领域
本发明涉及一种用于异氰酸酯聚合的催化剂,还涉及所述催化剂的制备方法,进一步涉及一种制备聚异氰酸酯的方法。
背景技术
聚异氰酸酯常规结构为三聚体和二聚体。三聚体即异氰酸酯单体三聚反应生成的六元环结构。该结构热稳定性好,尤其是脂肪族或脂环族的三聚体同时具有抗黄变的特性,但此结构粘度大;二聚体即异氰酸酯单体二聚反应生成,该结构稳定性弱于三聚体结构,但可降低体系粘度。
制备聚异氰酸酯的关键主要在于催化剂的选择,主要的催化剂包括有机碱金属和重金属、烷氨基膦、叔胺、季铵碱、季铵盐化合物及含氮的杂环化合物、吡啶类化合物等。
专利GB837120和US3211703提出将有机碱金属、重金属和叔胺作为制备聚异氰酸酯的催化剂,产品以三聚体结构为主,但在这些反应中金属类催化剂用量大,在使用过程中易析出导致产品发生浑浊;叔胺类催化剂在低温下反应活性过低,需要在较高温度下才保持较高活性,而且易导致产品产生氨味。
专利DE1150080提出将季铵碱作为制备聚异氰酸酯的催化剂,但在此催化剂的作用下反应过程容易发生飞温,导致反应过程无法控制,无法在生产中广泛应用。
专利US7001973披露了一种苄基取代的季铵羧酸盐催化剂,使用该催化剂催化反应得到了浅色的聚异氰酸酯,但以三聚体结构为主,并且反应中催化剂的用量较大,反应过程放热剧烈,不易控制。
专利US4912210涉及了吡啶类化合物和烷氨基膦催化剂,制备的产品以二聚体为主,基本无三聚体结构,且其单体转化率低。
专利US8097691提及了含膦的杂环化合物,可制备同时含三聚体和二聚体结构的产品,但催化剂制备过程复杂,低温下催化剂活性低。
因此,需要提供一种新型催化剂结构,与常规催化剂相比,催化剂制备过程简单,基于这种新型催化剂的聚异氰酸酯的制备方法,引发温度低,反应过程温和,保证较高单体转化率的前提下,获得含有三聚和二聚结构的聚异氰酸酯产品。
发明内容
本发明的目的是提供一种用于异氰酸酯聚合的催化剂及其制备方法,该催化剂能够用于制备含有三聚和二聚结构的聚异氰酸酯产品。所述催化剂引发温度低,反应过程温和,单体转化率高,聚异氰酸酯产品含有三聚和二聚结构。
本发明还提供一种使用所述催化剂制备聚异氰酸酯的方法,过程简单易行。
为达到以上技术目的,本发明采用如下技术方案:
一种用于异氰酸酯聚合的催化剂,其特征在于,所述催化剂的结构式为:
其中R1、R2、R3、R4相互独立的选自氢、苯基、具有1-10个碳原子的烷基、具有7-15个碳原子的烷苯基,优选氢、苯基、甲基、乙基、丙基、丁基、甲苯基、乙苯基;R5选自苯基、1-6个碳原子的烷基、1-6个碳原子的烷氧基、1-4个碳原子的烷氨基,优选苯基、甲基、乙基、正丙基、异丙基、甲氧基、乙氧基、二甲氨基、二乙氨基。
更优选的,所述催化剂选自结构式为式I、II、III、IV、V、VI的化合物中的一种或多种:
本发明所述的催化剂的制备方法,包括以下步骤:在无水无氧条件下,将摩尔比为1:1~1:1.5的N,N-二取代-1,2-二取代乙二胺与一取代二氯化膦在100-150℃加热混合,持续搅拌,在160-190℃下反应1-3h,得到浅黄色粘稠固体;经过重结晶得到目标产物1,3,2-二氮磷杂环戊烷类化合物。
制备催化剂的反应方程式如下:
本发明所述重结晶使用的溶剂可以使用本领域公知的合适的溶剂,优选乙酸乙酯和/或石油醚,更优选质量比为1:1的乙酸乙酯和石油醚的混合物。
本发明所述的N,N-二取代-1,2-二取代乙二胺优选自N,N-二甲基乙二胺、N,N-二乙基乙二胺、N,N-二苯基乙二胺、1,2-二苯基乙二胺、N,N-二甲基-1,2-二苯基乙二胺。
本发明所述的一取代二氯化膦优选自苯基二氯化膦、甲基二氯化膦、乙基二氯化膦、正丙基二氯化膦、异丙基二氯化膦、甲氧基二氯化膦、乙氧基二氯化膦、二甲氨基二氯化膦。
本发明所述的1,3,2-二氮磷杂环戊烷类化合物优选为N,N-二甲基-P-甲基-1,3,2-二氮磷杂环戊烷、N,N-二苯基-P-苯基-1,3,2-二氮磷杂环戊烷、P-甲氧基-4,5-二苯基-1,3,2-二氮磷杂环戊烷、N,N-二甲基-P-二甲氨基-4,5-二苯基-1,3,2-二氮磷杂环戊烷、N,N-二乙基-P-甲基-1,3,2-二氮磷杂环戊烷、N,N-二甲基-P-甲氧基-1,3,2-二氮磷杂环戊烷。
本发明所述的催化剂可用于催化制备聚异氰酸酯。
一种用本发明所述的催化剂制备聚异氰酸酯的方法,包括以下步骤:在惰性气体的保护下,将异氰酸酯原料在本发明所述催化剂的催化下进行自聚反应,反应温度为10-100℃,优选40-80℃。
适合本发明低聚方法的异氰酸酯原则上包括所有脂族异氰酸酯,其可以是单一一种脂族异氰酸酯或者彼此的混合物。以下列出的所有异氰酸酯的结构或构型异构体均为可选实例:双(异氰酸根合烷基)醚、丙烷二异氰酸酯、丁烷二异氰酸酯、戊烷二异氰酸酯、己烷二异氰酸酯(例如,六亚甲基二异氰酸酯,HDI)、庚烷二异氰酸酯、辛烷二异氰酸酯(例如,八亚甲基二异氰酸酯)、壬烷二异氰酸酯(例如,三甲基-HDI、TMDI,通常作为2,4,4-和2,2,4-异构体的混合物存在)和三异氰酸酯(例如,4-异氰酸根合甲基-1,8-辛烷二异氰酸酯)、癸烷二异氰酸酯(例如,十亚甲基二异氰酸酯)和三异氰酸酯、十一烷二异氰酸酯和三异氰酸酯、十二烷二异氰酸酯(例如,十二亚甲基二异氰酸酯)和三异氰酸酯、十四烷二异氰酸酯(例如,十四亚甲基二异氰酸酯)、1,4-环己烷二异氰酸酯(CHDI)、1,3-和1,4-双(异氰酸根合甲基)环己烷(H6XDI)、3-异氰酸根合甲基-3,5,5-三甲基环己基异氰酸酯(异佛尔酮二异氰酸酯,IPDI)、4,4’-二环己基甲烷二异氰酸酯(H12MDI)以及双(异氰酸根合甲基)降冰片烷(NBDI)、3(4)-异氰酸根合甲基-1-甲基-环己基异氰酸酯(IMCI)。优选采用HDI、TMDI、2-甲基戊烷-1,5-二异氰酸酯(MPDI)、H6XDI、NBDI、IPDI和H12MDI。
适合本发明低聚方法的异氰酸酯还可以包含芳香族异氰酸酯,其实例包含但不限于苯二亚甲基二异氰酸酯(XDI)、甲苯二异氰酸酯(TDI)。
按照本发明优选的是,作为低聚用的异氰酸酯优选是脂肪族二异氰酸酯和/或脂环族二异氰酸酯,更优选是戊基二异氰酸酯、六亚甲基二异氰酸酯(HDI)、八亚甲基二异氰酸酯、十亚甲基二异氰酸酯、十二亚甲基二异氰酸酯、十四亚甲基二异氰酸酯、1,4-环己烷二异氰酸酯(CHDI)、4,4’-二环己基甲烷二异氰酸酯(H12MDI)和异佛尔酮二异氰酸酯(IPDI)中的一种或多种。
本发明所述的催化剂的用量基于异氰酸酯原料的质量为100-1000ppm,优选300-600ppm。
反应过程中的引发温度即为异氰酸酯单体自聚反应明显加速,反应放热明显增加时的最低温度,实验过程中发现,该催化剂引发温度为40-50℃。
反应过程的温升大小可反映出反应过程热量释放的剧烈程度,同等条件下温升越大,热量释放剧烈,越不易控制。反应过程的温升测试方法:同样催化剂用量和反应温度下,加入催化剂后,不进行冷却控制,观察反应温度上升趋势,温升大小等于温度上升的最高温度减去起始温度,基于此催化剂的反应过程的温升为2-6℃。
反应液中-NCO含量基于反应液的重量达到23-25wt%时,可采用本领域公知的任意现有技术对反应进行终止,例如高温处理或加入催化剂毒物,所述高温处理的温度为120-150℃,处理时间为0.5-1.5h;所述催化剂毒物可选择酸和/或酸的衍生物,合适的例子包括但不限于磷酸、苯甲酰氯、苯甲磺酸酯、磷酸酯、亚磷酸酯、甲磺酸和对甲苯磺酸等中的一种或多种,催化剂毒物的加入量为与催化剂等摩尔量或者微过量。
将部分三聚后的反应液经过薄膜蒸发、萃取、精馏等工业常规方法脱除单体,获得的聚异氰酸酯产品的单体含量小于0.5wt%,基于无溶剂100%固含产品,采用液相法分析;二聚体含量40-50wt%、三聚体含量45-60wt%、五聚体以上含量低于5wt%,基于五聚体以上多聚体、三聚体和二聚体的总重,采用凝胶色谱法分析。
获得的100%固含无溶剂的聚异氰酸酯产品室温下为固体或者液体状态,可以直接使用或者用溶剂稀释后使用。所述溶剂可选自乙酸乙酯、乙酸丁酯、二甲苯、甲基异戊基酮和丙二醇甲醚醋酸酯等中的一种或多种,稀释后获得固含量为50-80wt%的产品,即100%固含无溶剂产品占比50-80wt%。
本文提及的催化剂可用于异氰酸酯的聚合催化,相对于常规催化剂,引发温度低,反应过程温和,单体转化率高,制备过程简单,聚异氰酸酯产品含有三聚和二聚结构。
具体实施方式
通过以下实施例将对本发明所提供的方法予以进一步的说明,但本发明并不因此而受到任何限制。
三聚体、二聚体、五聚体及以上多聚体含量采用凝胶色谱(色谱柱MZ-Gel SDplus10E3A,流动相四氢呋喃)测定;反应液和产品中的异氰酸酯单体含量采用液相法测试;催化剂结构采用NMR(Bruker DPX400)和质谱(安捷伦7890A-5975C)表征;产品粘度采用Brookfield DV-I Prime转子粘度计测试。
实施例1:
隔绝水分和氮气保护条件下,将1:1.2(摩尔比)的N,N-二甲基乙二胺与二氯甲基膦在反应瓶中混合,逐渐加热至110℃相互混合,持续搅拌,在170℃下反应1.5h,得到浅黄色粘稠液体,然后以质量比为1:1的乙酸乙酯和石油醚为溶剂重结晶分离得到1#催化剂,反应方程式如下:
1#催化剂表征数据如下:
1HNMR(400M,TMS):δ2.67(t,4H),2.43(s,6H),1.18(s,3H)
13CNMR(100M,TMS):δ57.8,38.6,10.3
[M+H]⊕:133.0889(ESI)
实施例2:
隔绝水分和氮气保护条件下,将1:1.1(摩尔比)的N,N-二乙基乙二胺与二氯甲基膦在反应瓶中混合,逐渐加热至130℃相互混合,持续搅拌,在180℃下反应3h,得到浅黄色粘稠液体,然后以质量比为1:1的乙酸乙酯和石油醚为溶剂重结晶分离得到2#催化剂,反应方程式如下:
2#催化剂表征数据如下:
1HNMR(400M,TMS):δ2.96(t,4H),2.67(s,4H),1.18(s,3H),
1.02(s,6H)
13CNMR(100M,TMS):δ55.6,43.3,14.4,10.9
[M+H]⊕:161.1129(ESI)
实施例3:
隔绝水分和氮气保护条件下,将1:1.3(摩尔比)的N,N-二甲基乙二胺与二氯甲氧基膦在反应瓶中混合,逐渐加热至140℃相互混合,持续搅拌,在180℃下反应2.5h,得到浅黄色粘稠液体,然后以质量比为1:1的乙酸乙酯和石油醚为溶剂重结晶分离得到3#催化剂,反应方程式如下:
3#催化剂表征数据如下:
1HNMR(400M,TMS):δ3.51(s,3H),2.67(s,4H),2.43(s,6H),
13CNMR(100M,TMS):δ58.9,54.4,37.4
[M+H]⊕:149.0765(ESI)
实施例4:
隔绝水分和氮气保护条件下,将1:1(摩尔比)的N,N-二苯基乙二胺与二氯苯基膦在反应瓶中混合,逐渐加热至120℃相互混合,持续搅拌,在180℃下反应2.5h,得到浅黄色粘稠固体,然后以质量比为1:1的乙酸乙酯和石油醚为溶剂重结晶得到4#催化剂,反应方程式如下:
4#催化剂表征数据如下:
1HNMR(400M,TMS):δ7.45(s,3H),7.38(s,2H),7.23(s,4H),6.77(s,2H),6.60(s,4H),3.29(s,4H)
13CNMR(100M,TMS):δ147.5,141.7,131.0,129.5,128.7,120.8,114.8,58.5
[M+H]⊕:319.1359(ESI)
实施例5:
隔绝水分和氮气保护条件下,将1:1.5(摩尔比)的1,2-二苯基乙二胺与二氯甲氧基膦在反应瓶中混合,逐渐加热至130℃相互混合,持续搅拌,在180℃下反应2h,得到浅黄色粘稠固体,然后以质量比为1:1的乙酸乙酯和石油醚为溶剂经过重结晶得到5#催化剂,反应方程式如下:
5#催化剂表征数据如下:
1HNMR(400M,TMS):δ7.40(s,4H),7.28(s,6H),4.52(s,2H),3.51(s,3H),2.0(s,2H)
13CNMR(100M,TMS):δ143.5,128.5,126.8,68.1,58.3
[M+H]⊕:273.1151(ESI)
实施例6:
隔绝水分和氮气保护条件下,将1:1.3(摩尔比)的N,N-二甲基1,2-二苯基乙二胺与二氯(二甲氨基)膦在反应瓶中混合,逐渐加热至125℃相互混合,持续搅拌,在180℃下反应2h,得到浅黄色粘稠固体,然后以质量比为1:1的乙酸乙酯和石油醚为溶剂经过重结晶得到6#催化剂,反应方程式如下:
6#催化剂表征数据如下:
1HNMR(400M,TMS):δ7.40(s,4H),7.28(s,6H),4.52(s,2H),2.43(t,12H)。
13CNMR(100M,TMS):δ138.3,128.5,127.9,127.0,76.4,39.4,34.3。
[M+H]⊕:314.1781(ESI)
实施例7~12:
将800g IPDI置于装有回流冷凝管、搅拌器、温度计和氮气入口的圆底烧瓶中,加热至60℃,然后分别加入1#-6#催化剂,并不断搅拌,反应过程中呈现升温,控制反应温度在60-80℃之间,反应液的-NCO值达到23-24wt%之间时,以反应液的重量为基准计算,立即加入与初始催化剂等摩尔量的苯甲酰氯,继续搅拌15min即可终止反应。
使用薄膜蒸发器在温度180℃,绝对压力低于200Pa的条件下蒸发脱除部分三聚反应液中的单体,获得100%固含无溶剂产品,其单体含量低于0.5wt%,得到的100%固含无溶剂产品溶解于乙酸丁酯中,可得固含量70wt%的产品,然后测试五聚体及以上多聚体、三聚体、二聚体、单体含量及产品粘度。
反应条件及反应结果见表1。
对比例1~2:
催化剂分别采用三(二甲胺基)膦(即7#催化剂)和四丁基氢氧化铵(即8#催化剂),其余参照实施例7。反应条件及反应结果见表1。
表1实施例7~12及对比例1~2反应条件及反应结果
说明:此处的五聚体以上多聚体、三聚体、二聚体含量采用凝胶色谱测试,对此三种组成进行面积归一化计算,不包括单体含量,基于产品中的固体产物(五聚体以上多聚体、三聚体和二聚体);单体含量测试采用液相法,基于100%固含无溶剂产品。
8#催化剂制备产品五聚体以上含量40%,表中结果可知,基于IPDI,获得的产品与常规技术产品相比,粘度在常规二聚体和常规三聚体产品之间,组成兼顾了常规产品三聚体和二聚体主体的结构。
实施例13~18:
将1000g HDI置于装有回流冷凝管、搅拌器、温度计和氮气入口的圆底烧瓶中,加热至35℃,分别加入催化剂1#-6#并不断搅拌,反应过程中呈现升温,控制反应温度在45-55℃之间,反应液的NCO值达到24-25wt%之间时,以反应液的重量为基准计算,立即加入与催化剂等摩尔量的磷酸,继续搅拌15min即可终止反应。
使用薄膜蒸发器在温度150℃,绝对压力低于100Pa的条件下蒸发脱除部分三聚反应液中的单体,获得100%固含无溶剂产品,其单体含量低于0.5wt%,即得到所需产品,然后测试五聚体及以上多聚体、三聚体、二聚体、单体含量及产品粘度。
反应条件及反应结果见表2。
对比例3~4:
催化剂分别采用三(二甲胺基)膦(即7#催化剂)和四丁基氢氧化铵(即8#催化剂),其余参照实施例13。反应条件及结果见表2。
表2实施例13~18及对比例3~4反应条件及反应结果
说明:此处的五聚体以上多聚体、三聚体、二聚体含量采用凝胶色谱测试,对此三种组成进行面积归一化计算,不包括单体含量,基于产品中的固体产物(五聚体以上多聚体、三聚体和二聚体);单体含量测试采用液相法,基于100%固含无溶剂产品。
8#催化剂制备产品五聚体以上含量45%,表中结果可知,基于HDI,获得的产品粘度在常规二聚体和常规三聚体产品之间,组成兼顾了常规产品的三聚体和二聚体主体结构。
一种异氰酸酯聚合催化剂及其制备方法,及其用于制备聚异氰酸酯的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。










动态评分
0.0