

IPC分类号 : A61K31/517,A61K31/53,A61K31/136,C07D211/00,C07D239/70







专利摘要
本发明涉及用于抑制Akt/PKB通路的物质和方法。在一个实施方案中,本发明的化合物抑制Akt蛋白的激酶活性和/或磷酸化水平。本发明还涉及用于抑制或杀灭癌细胞或其它其中Akt蛋白的表达升高或组成型活化的细胞的方法,该方法包括使所述细胞接触有效量的式I的化合物。本发明还涉及用于治疗人或动物癌症或肿瘤的方法,该方法包括对所述人或动物给予有效量的式I的化合物。
说明书
技术领域具有抗肿瘤活性的AKT/PKB抑制剂
技术背景背景技术
Akt,也称作蛋白激酶B,代表了丝氨酸/苏氨酸激酶亚族。Akt首先被描述为v-akt癌基因产物的细胞同源物(Bellacosa等,1991),并且它具有三个成员Akt1/PKBα,Akt2/PKBβ和Akt3/PKBγ(Cheng等,1992;Jones等,1991a;Jones等,1991b)。Akt的活化依赖于介导其膜转位的普列克底物蛋白同源性(PH)结构域的完整性和活化环中的Thr308和Ser473的磷酸化(Konishi等,1995)。由PI3K产生的磷肌醇类PtdIns-3,4-P2和PtdIns-3,4,5-P3直接结合Akt的PH结构域,从而驱动分子的构象改变,这能够使Ak t的活化环被Thr308上的PDK1磷酸化(Datta等,1999)。Akt的完全活化还与C-末端疏水性基序内的Ser473磷酸化相关(Datta等,1999)。尽管PDK1在Thr308磷酸化中的作用得到充分确立,但是对Ser473磷酸化的机理存在争论。已经提出了负责这一改变的大量候选酶,包括整联蛋白-连接的激酶(Persad等,2001)、在与激酶PRK2复合时的PDK1(Wiek等,2000)、通过自磷酸化的Akt自身(Toker等,2000)、DNA-依赖性激酶(Feng等,2004)和rictor-mTOR复合物(Sarbassoy等,2005)。Akt的活性受到肿瘤抑制基因PTEN负调节,它通常在人恶性肿瘤中突变(Vazquez等,2000)。PTEN编码双向特异性蛋白和通过将它们转化成PtdIns-4,5-P2而降低细胞内PtdIns-3,4,5-P3水平的脂质磷酸酶,由此抑制PI3K/Akt通路(Stambolic等,1998)。
Akt使发挥其正常细胞功能的大量分子磷酸化和/或与它们发生相互作用,包括在细胞增殖、存活、迁移和分化中的作用(Cheng等,2001)。多项证据表明Akt是肿瘤发生和发展中的关键参与者。此外,在至多50%的所有人肿瘤中检测到Akt通路异常过度活化(Sun等,2001;Cheng等,1997)并且与化学抗性紧密相关(West等,2002)。因此,Akt已经成为抗癌药研发的有吸引力的靶标(West等,2002)。
在过去的几年中,通过组合化学、高流通量和实际筛选,鉴定了Akt通路的12个抑制剂。首先研发了Akt的基于脂质的抑制剂,包括哌立福辛(Kondapaka等,2003),PX-316(Meuillet等,2004)和磷脂酰肌醇醚脂质类似物(Castillo等,2004),它们被设计成与Akt的PH结构域发生相互作用。此外,已经使用化学文库的高流通量筛选和合理设计鉴定了几种Akt拮抗剂。这些抑制剂包括醋酸9-甲氧基-2-甲基椭圆玫瑰树碱(Jin等,2004),吲唑-吡啶A-443654(Luo等,2005),亚型-特异性变构激酶抑制剂(Lindsley等,2005)和Akt/PKB信号传导抑制剂-2(API-2),也称作曲西立滨/TCN(Yang等,2004)。API-2/TCN是在预先进行的I期和II期试验中表现出抗肿瘤活性的三环核苷,但存在多种毒性,包括肝毒性,高血糖症,血小板减少症和高甘油三酯血症,从而排除了进一步的研发(Feun等,1993;Hoffman等,1996)。通过筛选NCI多样性组,我们预先证实API-2抑制Akt激酶活性并且刺激表现出高Akt活性的人癌细胞异种移植物的细胞凋亡(Yang等,2004)。这一发现在研究该药中提供了新的关注并且提高了降低的剂量可以抑制Akt和诱导肿瘤细胞细胞凋亡、而无预先相关的副作用的可能性(Yang等,2004;Cheng等,2005)。
发明内容发明内容
本发明涉及用于抑制Akt/PKB通路的化合物、组合物和方法。在一个实施方案中,本发明的化合物抑制Akt蛋白的激酶活性和/或磷酸化水平。本发明的化合物具有式I中所示的一般结构。在一个具体的实施方案中,本发明的化合物(本文称作API-1)具有如下结构:
本发明的化合物,诸如API-1抑制具有异常Akt的人肿瘤细胞中的Akt信号传导,从而导致细胞生长抑制和诱导细胞凋亡。在异种移植裸鼠模型中,本发明的化合物明显抑制具有过度活化的Akt的细胞中的肿瘤生长,但在具有低水平Akt的肿瘤中则没有抑制效果。
本发明还涉及用于抑制或杀伤癌细胞或其它其中Akt蛋白表达升高或组成型活化的细胞的方法,包括使所述细胞接触有效量的式I的化合物。
本发明还涉及用于治疗人或动物癌症或肿瘤的方法,包括对所述人或动物给予有效量的式I的化合物。
丝氨酸/苏氨酸激酶Akt/PKB通路通常在人癌症中过度活化并且作为转导细胞内和细胞外癌基因信号的主要节点起作用,且由此它提供了分子治疗剂的靶标。通过筛选National Cancer InstituteDiversity Set(美国国家癌症研究所(NCI)的多样性组),鉴定了小分子Akt通路抑制剂API(Akt/PKB信号传导抑制剂)-1。API-1抑制Akt家族的三个成员的激酶活性和磷酸化水平。然而,它对上游激活物PI3K和PDK1的活性没有影响。此外,组成型活性的Akt的激酶活性和磷酸化水平在细胞培养物中受到API-1显著地抑制,而它对体外的Akt激酶活性无影响。API-1对Akt具有高度选择性并且不抑制PKC,SGK,PKA,STAT3,Erk-1/2或JNK活化。API-1抑制Akt导致在隐含组成性激活的Akt的人癌细胞中诱导细胞生长停止和细胞凋亡。显然,API-1选择性抑制其中Akt升高的人癌细胞在裸鼠中的肿瘤生长,而在Akt不升高的那些癌细胞中则不抑制。这些数据提示API-1是具有体外和体内抗肿瘤活性的Akt通路抑制剂并且可以成为具有表达过度活化的Akt的癌症的患者的潜在抗癌药。
Akt是主要的调节癌细胞存活、生长和肿瘤发展的通路。已经充分记录了Akt激酶水平升高促成了对各种癌症疗法的抗性,包括细胞毒性化疗药以及Bcr-Abl(Gleevee)、Her2/Ncu(Hercptin)和mTOR(雷帕霉素)的小分子抑制剂。阻断Akt抑制了具有过度活化的Akt的癌细胞中的肿瘤生长并且使癌细胞对化疗和其它靶向治疗更敏感。API-1与其它抗肿瘤药的联合用药提供了更有效的抗肿瘤作用。
附图说明附图说明
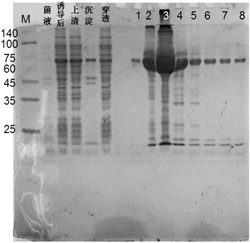
图1A-1D是来自NCI Diversity Set的将API-1鉴定为候选的Akt抑制剂的图解表示法。图1A表示API-1的化学结构。图1B是表示API-1抑制Akt的三个成员的示意图。用HA-Akt1,-AKT2和-AKT3转染HEK293细胞并且在EGF刺激前用API-1(10μM)处理,裂解细胞并且用抗-HA抗体免疫沉淀。对免疫沉淀物进行体外激酶测定(上图)。下图是表示使用抗-HA抗体检测的转染的Akt1,AKT2和AKT3的表达的蛋白质印迹。图1C是表示API-1抑制表达过度活化的Akt的OVCAR3细胞中Akt磷酸化水平的示意图。使用API-1在所示浓度下处理细胞2小时并且使用抗-磷酸-Akt-S473抗体进行免疫印迹分析(上图)。下图表示总Akt的表达。图1D是表示API-1不抑制体外Akt的示意图。含所示量API-1的激酶缓冲液中的重组组成型活性Akt蛋白的体外激酶测定。化合物E,即多种激酶抑制剂ATP-竞争者用作阳性对照。将本实验重复三次。
图2A-2G是表示API-1不抑制PI3K,PDK1和AGC激酶家族的紧密相关的成员的示意图。图2A是表示体外PI3K激酶测定的示意图。使HEK293细胞血清饥饿并且在EGF刺激前用API-1(10μM)或渥曼青霉素(1nM)处理30分钟。裂解细胞并且用抗-p110α抗体免疫沉淀。使用PI-4-P作为底物对免疫沉淀物进行体外激酶测定。图2B是表示API-1对PDK1活化作用的示意图。使用PDK1激酶试剂盒(UpstateBiotechnology Inc),根据制造商的说明,在所示化合物存在下进行体外激酶测定。图2C是表示体外PKA激酶测定的示意图。在含所示抑制剂(API-1或PKAI)和底物肯普肽的ADB缓冲液(UpstateBiotechnology Inc)中孵育重组PKA。对激酶活性进行定量测定。图2D是表示API-1对活细胞中PKA,PKC和PDK激酶活性的作用的示意图。用所示浓度的API-1将OVCAR3细胞处理1小时。裂解细胞并且用所示抗体免疫印迹。图2E是表示体外SGK激酶测定的示意图。将重组SGK与API-1或化合物E一起孵育。通过添加SGK底物肽和[γ-32P]ATP来启动激酶测定。对激酶活性进行定量测定。图2F是表示用HA-SGK转染HEK293细胞并且在EGF刺激前用API-1或渥曼青霉素处理时的结果的示意图。使用HA-SGK免疫沉淀物、应用组蛋白-H2B作为底物进行体外激酶测定。图2G是表示API-1不抑制Erk,p38,JNK和Stat3磷酸化的示意图。用API-1将OVCAR3细胞处理3小时并且用所示抗体免疫印迹。
图3A-3D是表示API-1抑制组成型活性Akt及其下游靶标的示意图。图3A是表示API-1抑制组成型活性Akt的示意图。用所示HA-myr-Akt1,HA-Akt1-E40K和HA-myr-Akt2转染HEK293细胞。在用API-1处理1小时后,裂解细胞并且用抗-HA抗体免疫沉淀。对免疫沉淀物进行体外激酶测定(上图)并且用所示抗体免疫印迹(中间图和下图)。图3B是表示API-1抑制Akt下游靶标磷酸化的示意图。用API-1(10μM)将OVCAR3细胞处理所示次数并且用所示抗体免疫印迹。API-1显著降低了Akt及其下游靶标GSK3β和S6蛋白的磷酸化水平。在图3C中,AKT-SI1抑制Akt下游靶标磷酸化。在图3D中,AKT-SI1不干扰mTORC1和mTORC2复合物。
图4A-4G是表示API-1抑制Akt活性和细胞生长并且诱导具有升高Akt的人癌细胞中细胞凋亡的示意图。图4A是表示使用API-1处理后的结果的蛋白质印迹,使用抗-磷酸-Akt-T308和裂解的PARP抗体在所示人癌细胞系中检测Akt磷酸化水平和PARP裂解(上图和中间图)。使用抗-肌动蛋白抗体再探测印迹(下图)。图4B和4C是细胞增殖测定,其中用不同剂量的API-1将所示细胞系处理24小时然后用MTT测定法分析。图4D-4G表示细胞凋亡分析,其中用API-1处理细胞并且用膜联蛋白V和PI染色且通过FACScan分析。
图5A-5L是表示API-1在小鼠异种移植物中具有升高Akt的癌细胞系中表现出抗肿瘤活性的示意图。图5A-5F是对照组(图5A)和API-1-处理组(图5B)的照片和相关示意图(图5C-5F)。将肿瘤细胞经皮下注入具有低水平Akt细胞的裸鼠右侧和具有升高水平Akt细胞的裸鼠左侧。当肿瘤达到约100-150mm3平均大小时,用媒介物或10mg/kg/天API-1如“材料和方法”中所示处理动物。用表达升高水平的Akt的PANC1/OVCAR3(分别为图5E和5C)和显示出低水平Akt的COLO357/OVCAR5(分别为图5F和5D),用API-1或媒介物(A)处理的异种移植物处理小鼠的表示。B组表示使用10只小鼠/组的肿瘤生长曲线。图5G-5J表示实验结束时肿瘤大小(左)和重量(右)的实例。API-1比DMSO对照组显著降低PANC1和OVCAR3异种移植物中的肿瘤重量(*P=0.02)。图5K和5L表明API-1抑制体内Akt磷酸化。裂解API-1处理和未处理的肿瘤样本并且使用所示抗体免疫印迹。
图6A-6C表示AKT-SI1抑制IGF-1诱导的Akt质膜转位。在盖玻片上用Myc-AKT1转染HeLa细胞,血清饥饿过夜且然后用(图6C)或不用(图6B)AKT-SI1处理30min,然后用IGF1刺激15分钟。固定后,用抗-Myc单克隆抗体免疫染色细胞,随后用FITC-缀合的二次抗体染色细胞以揭示出在胞质溶胶或膜中存在表位-标记的蛋白质(图6B)。将不用AKT-SI1和IGF1处理的细胞用作对照组(图6A)。
图7A-7E表示在Akt激酶活性,尤其是组成型活性Akt的抑制中AKT-SI1比API-2/TCN更有效。尽管AKT-SI1结构与API-2/TCN共有核糖的糖部分(图7A),但是这2种分子的剩余部分不具有化学相似性。然而,我们比较了它们抑制Akt的能力。用野生型Myc-AKT1(图7B和7C)和组成型活性Myc-AKT1-E17K(图7D和7E)转染HEK293细胞。在36h孵育后,使细胞血清饥饿过夜。用AKT-SI1(图7B和7D)或API-2/TCN(图7C和7E)将野生型Akt-转染细胞处理30min且随后用EGF刺激15min。使用抗-Myc抗体进行免疫沉淀并且使用组蛋白H2B作为底物对免疫沉淀物进行体外激酶测定(上图)。对AKT-SI1和API-2/TCN抑制Akt激酶活性进行定量测定并且计算为相对活性(中图)。蛋白质印迹分析显示了免疫沉淀的AKT1蛋白(下图)。将实验重复三次。
图8A和8B表示AKT-SI1和API-2/TCN抑制磷酸-Akt水平。用所示试剂处理野生型Myc-AKT1和组成型活化Myc-AKT1-E17K转染的HEK293细胞并且用抗-磷酸-Akt-S473(上图)和-Myc(下图)抗体进行免疫印迹。
具体实施方式具体实施方式
本发明涉及抑制Akt/PKB通路的化合物和组合物。在一个实施方案中,本发明的化合物抑制Akt蛋白的激酶活性和/或磷酸化水平。本发明的化合物具有式I所示的一般结构:
其中
X独立地是O,N或S;
Y是O,N或S;
R1独立地是-H,-OH,-NH2,-NO2,卤素,任选被-OH取代的烷基或任选被-OH取代的烷氧基;且
R2是-H,-OH,-NH2,-C(O)NH2,烷基或烷氧基,它们中的任一个可以任选被-OH,卤素,烷基或烷氧基取代;
R3是-H,-OH,-NH2,-C(O)NH2,烷基或烷氧基,它们中的任一个可以任选被-OH,卤素,烷基或烷氧基取代;或
其类似物或药学上可接受的盐。
在一个实施方案中,X各自是N。在一个典型的实施方案中,Y是O。在另一个典型的实施方案中,R1各自独立地是-OH或-CH2OH。在另一个实施方案中,R2是任选被-OH取代的-NH2。在另一个实施方案中,R2是-C(O)NH2。
在一个具体的实施方案中,本发明的化合物具有如下结构(式II):
本发明的化合物不仅抑制因诸如PTEN突变这类上游调节物改变而产生的高度活化的野生型Akt,而且抑制组成型活性Akt突变体,包括myr-AKT1,Myr-AKT2和E40K-AKT1。近期研究鉴定了人乳腺、结肠直肠和卵巢癌中AKT1的PH结构域中的复发的体细胞突变,它导致脂质结合袋中第17位氨基酸(E17K)上的谷氨酸发生赖氨酸取代(Carpten等,2007)。Lys 17改变所述袋的静电相互作用并且与磷酸肌醇配体形成新的氢键。这种突变在小鼠中通过病理性局限于质膜而活化AKT1,转化细胞并且诱导白血病。此外,E17K取代降低了对变构激酶抑制剂的敏感性(Carpten等,2007)。因为E40K-AKT1突变体与可局限于质膜的E17K类似(Bellacosa等,1998),所以API-1也可以抑制E17K-AKT1。
尽管可以将本发明的化合物作为单独的化合物给药,但是还可以将这些化合物作为药物组合物的组成部分给药。本发明由此进一步提供了混有至少一种药学上可接受的载体的组合物。该药物组合物适合于各种给药通路,诸如肠、胃肠外、静脉内、局部、皮下等。给药可以是连续的或在不同间隔,正如本领域技术人员可以确定的。
可以根据制备药学上有用的组合物的公知方法配制本发明的化合物。制剂描述在本领域技术人员众所周知和易于得到的许多来源中。例如,Remington’s Pharmaceutical Science(Martin 1995)中描述了可以用于本发明的制剂。适合于给药的制剂包括,例如无菌注射水溶液,它可以包含抗氧化剂、缓冲剂、抑菌剂和使制剂与指定接受者血液等渗的溶质;和可以包括助悬剂和增稠剂的无菌水和非水混悬液。可以将制剂提供在单位剂量或多剂量容器中,例如密封安瓿和小瓶,并且可以在使用前在仅需要无菌液体载体例如注射用水条件的冷冻干燥(冻干)条件下储存。可以由无菌粉末、颗粒、片剂等制备临时注射溶液和混悬液。应理解除上述具体描述的成分外,本发明的组合物还可以包括本领域在所示制剂类型方面常规的其它试剂。
本文所用的烷基意旨具有1至20个碳原子的直链或支链饱和或单-或多不饱和烃基,并且C1-X烷基意旨含至多“X”个碳原子数的直链或支链烷基。例如,C1-6烷基意旨含至多6个碳原子的直链或支链烷基。烷氧基意旨烷基-O-基团,其中烷基如上所述。
术语“卤素”意旨周期表中的卤素,诸如氟、氯、溴或碘。
术语“任选取代的”意旨任选在任一可利用的位置或多个位置上被一个或多个有机或无机基团(例如烷基、芳基、杂芳基、酰基、烯基、环烷基、杂环烷基、环烯基、杂环烯基或卤素)取代。
饱和烷基的实例包括,但不限于甲基,乙基,N-丙基,异丙基,N-丁基,叔丁基,异丁基,仲-丁基,N-戊基,N-己基,N-庚基和N-辛基。不饱和烷基是具有一个或多个双键或三键的基团。不饱和烷基包括,例如乙烯基,丙烯基,丁烯基,己烯基,乙烯基,2-丙炔基,2-异戊烯基,2-丁二烯基,乙炔基,1-丙炔基,3-丙炔基和3-丁炔基。特别地,“烷基”可以包括,例如甲基,乙基,丙基,异丙基,丁基,异丁基,仲-丁基,戊基,3-戊基,己基,庚基,辛基,壬基,癸基,十一烷基,十二烷基,十三烷基,十四烷基,十五烷基,乙烯基,1-丙烯基,2-丙烯基,1-丁烯基,2-丁烯基,3-丁烯基,1-戊烯基,2-戊烯基,3-戊烯基,4-戊烯基,1-己烯基,2-己烯基,3-己烯基,4-己烯基,5-己烯基,1-庚烯基,2-庚烯基,3-庚烯基,4-庚烯基,5-庚烯基,1-壬烯基,2-壬烯基,3-壬烯基,4-壬烯基,5-壬烯基,6-壬烯基,7-壬烯基,8-壬烯基,1-癸烯基,2-癸烯基,3-癸烯基,4-癸烯基,5-癸烯基,6-癸烯基,7-癸烯基,8-癸烯基,9-癸烯基;1-十一碳烯基,2-十一碳烯基,3-十一碳烯基,4-十一碳烯基,5-十一碳烯基,6-十一碳烯基,7-十一碳烯基,8-十一碳烯基,9-十一碳烯基,10-十一碳烯基,1-十二碳烯基,2-十二碳烯基,3-十二碳烯基,4-十二碳烯基,5-十二碳烯基,6-十二碳烯基,7-十二碳烯基,8-十二碳烯基,9-十二碳烯基,10-十二碳烯基,11-十二碳烯基,1-十三碳烯基,2-十三碳烯基,3-十三碳烯基,4-十三碳烯基,5-十三碳烯基,6-十三碳烯基,7-十三碳烯基,8-十三碳烯基,9-十三碳烯基,10-十三碳烯基,11-十三碳烯基,12-十三碳烯基,1-十四碳烯基,2-十四碳烯基,3-十四碳烯基,4-十四碳烯基,5-十四碳烯基,6-十四碳烯基,7-十四碳烯基,8-十四碳烯基,9-十四碳烯基,10-十四碳烯基,11-十四碳烯基,12-十四碳烯基,13-十四碳烯基,1-十五碳烯基,2-十五碳烯基,3-十五碳烯基,4-十五碳烯基,5-十五碳烯基,6-十五碳烯基,7-十五碳烯基,8-十五碳烯基,9-十五碳烯基,10-十五碳烯基,11-十五碳烯基,12-十五碳烯基,13-十五碳烯基或14-十五碳烯基;“烷氧基”可以包括甲氧基,乙氧基,丙氧基,异丙氧基,丁氧基,异丁氧基,仲丁氧基,戊氧基,3-戊氧基,己氧基,庚氧基,辛氧基,壬氧基,癸氧基,十一氧基,十二烷氧基,十三烷氧基,十四烷氧基或十五氧基。
本发明的化合物包括本领域技术人员可以制备的所有水合物和盐。在本发明的化合物的碱性或酸性足以形成稳定的无毒性酸或碱式盐的条件下,将化合物作为盐给予可能是适当的。药学上可接受的盐的实例是与形成生理学可接受的阴离子的酸形成的有机酸加成的盐,例如甲苯磺酸盐、甲磺酸盐、乙酸盐、柠檬酸盐、丙二酸盐、酒石酸盐、琥珀酸盐、苯甲酸盐、抗坏血酸盐、α-酮戊二酸盐和α-甘油磷酸盐。还可以形成合适的无机盐,包括盐酸盐、硫酸盐、硝酸盐、碳酸氢盐和碳酸盐。
可以使用本领域众所周知的标准方法获得化合物的药学上可接受的盐,例如通过使足够碱性的化合物,诸如胺与合适的酸反应得到生理学可接受的阴离子。还可以制备羧酸的碱金属(例如钠、钾或锂)或碱土金属(例如钙)盐。
本文所用的术语“类似物”意旨基本上与另一种化合物相同、但可以通过例如添加酸性基团、氧化或还原母体结构来修饰的化合物。易于使用通常已知的标准反应制备本发明化合物的类似物。这些标准反应包括,但不限于氢化、烷基化、乙酰化和酸化反应。本领域技术人员可以通过使用科学文献中已知的标准方法保护所有存在于分子上的官能基并且在进行所需反应后使其脱保护来进行化学修饰(Greene等,1999;Honda等,1997;Honda等,1998;Konoike等,1997;Honda等,2000;将这些文献各自完整地引入本文作为参考)。可以使用细胞测定法或者其它体外或体内测定法鉴定或确认表现出所需生物活性(诸如诱导细胞凋亡,细胞毒性,细胞稳定性(cytostaticity),诱导细胞周期停止,抗血管生成特性等)的类似物。
可以理解本发明的化合物可以包含一个或多个不对称取代的碳原(即碳中心)。在本发明化合物上存在一个或多个不对称中心可以产生立体异构体,并且在每种情况中,应理解本发明可以扩展至所有这类立体异构体,包括对映体和非对映异构体及其混合物(包括外消旋混合物)。
本发明的化合物和组合物用于各种非治疗和治疗目的。所述化合物和组合物可以用于减少动物和人的异常细胞生长。由于化合物的这类抗增殖特性,所以它们用于在体外和体内减少各种环境中不需要的细胞生长。
可以通过任一合适的治疗方法和目前或预期本领域技术人员公知的技术治疗性施用化合物和包含它们的组合物。此外,本发明的化合物具有作为制备其它有用的化合物和组合物的原料物质或中间体的用途。
可以将本发明的化合物及其任选与药学上可接受的载体,诸如惰性稀释剂组合的组合物在一个或多个解剖部位上局部给药,诸如不需要的细胞生长的部位(诸如肿瘤部位或良性皮肤生长,例如注射或局部施用于肿瘤或皮肤生长)或真菌感染部位。可以通过全身给予,诸如通过静脉内或口服给予本发明的化合物及其任选与药学上可接受的载体,诸如惰性稀释剂或可同化的可食用口服递送用载体组合的组合物。可以将它们包封在硬或软胶囊内,可以将它们压制成片剂或直接与患者膳食的食物掺合。就口服治疗给药而言,可以将活性化合物与一种或多种赋形剂合并并且以可摄入的片剂、含片、糖锭、胶囊、酏剂、混悬液、糖浆剂、糯米纸囊剂、气雾喷雾剂等形式使用。
片剂、锭剂、丸剂、胶囊等还可以包含如下成分:粘合剂,诸如黄蓍胶,阿拉伯胶,玉米淀粉或明胶;赋形剂,诸如磷酸二钙;崩解剂,诸如玉米淀粉,马铃薯淀粉,藻酸等;润滑剂,诸如硬脂酸镁;和甜味剂,诸如蔗糖,果糖,乳糖或阿司帕坦或可以加入矫味剂,诸如薄荷,冬青油或樱桃香料。当单位剂型是胶囊时,它除可以包含上述类型物质外还可以包含液体载体,诸如植物油或聚乙二醇。可以存在各种其它材料作为包衣衣料,或用于改变固体单位剂型的物理形式。例如,可以用明胶、蜡、虫胶或糖等给片剂、丸剂或胶囊包衣。糖浆剂或酏剂可以包含活性化合物、蔗糖或果糖(作为甜味剂),对羟基苯甲酸甲酯和对羟基苯甲酸丙酯(作为防腐剂),染料和矫味剂(诸如樱桃或橙香料)。当然,用于制备任一单位剂型的任何材料均应是药学上可接受的并且使用基本上无毒性的用量。此外,可以将活性化合物掺入缓释制剂和装置。
可以通过输注或注射经静脉内、肌内或腹膜内给予本发明的化合物和组合物,包括其药学上可接受的盐或类似物。可以用水(任选混合了无毒性表面活性剂)制备活性剂或其盐的溶液。还可以用甘油、液体聚乙二醇、三醋精及其混合物和用油制备混悬液。在通常的储存和使用条件下,这些制剂可以包含防腐剂以防止微生物生长。
适合于注射或输注的药物剂型可以包括无菌水溶液或分散液或包含活性成分的适合于临时制备无菌可注射或可输注溶液或分散液的任选包囊在脂质体中的无菌粉末。最终剂型在制备和储存条件下应是无菌的,流动的和稳定的。液体载体或媒介物可以是溶剂或液体分散介质,包括,例如水、乙醇、多元醇(例如甘油、丙二醇、液体聚乙二醇等)、植物油、无毒性甘油酯类及其合适的混合物。例如,可以通过形成脂质体,通过维持分散体中所需的颗粒大小或通过使用表面活性剂来维持适当的流动性。任选可以用各种其它抗菌和抗真菌剂,例如对羟基苯甲酸酯类、三氯叔丁醇、苯酚、山梨酸、硫柳汞等来防止微生物的作用。在许多情况中,优选包括等渗剂,例如糖类,缓冲剂或氯化钠。可以通过包含延缓吸收的试剂,例如单硬脂酸铝和明胶来延长可注射组合物的吸收。
通过将所需量的本发明化合物掺入含有各种其它上述成分的适当的溶剂,如果需要的话过滤灭菌来制备无菌可注射溶液。就制备无菌可注射溶液的无菌粉末而言,优选的制备方法是真空干燥和冷冻干燥技术,它们产生活性成分与存在于上述无菌过滤的溶液中的任一额外所需成分的粉末。
就局部给药而言,可以将本发明的化合物作为液体或固体施用。然而,一般需要以局部方式将它们作为与皮肤可接受的可以为固体或液体的载体组合的组合物来施用于皮肤。可以将本发明的化合物和组合物局部施用于受试者皮肤以减小恶性或良性生长的大小(并且可以包括完全除去)或治疗感染部位。可以将本发明的化合物直接施用于生长或感染部位。优选将化合物以制剂形式施用于生长或感染部位,诸如以软膏剂、霜剂、洗剂、溶液、酊剂等形式。还可以使用用于给表皮损害递送药理学物质的递药系统,诸如描述在美国专利US 5,167,649中。
有用的固体载体包括固体细粉,诸如滑石粉、粘土、微晶纤维素、二氧化硅、氧化铝等。有用的液体载体包括水、醇类或二醇类或水-醇/二醇掺合物,其中可以将化合物以有效水平任选借助于无毒性表面活性剂溶解或分散。可以加入佐剂,诸如香料和额外的抗微生物剂以优化指定应用的特性。可以从吸收垫中施用所得液体组合物,用于浸渍绷带和其它包扎或例如使用泵型或气雾型喷雾器喷在受侵害区域。
还可以将增稠剂,诸如合成聚合物、脂肪酸、脂肪酸盐和酯类、脂肪醇类、改性纤维素或改性矿物材料与液体载体联用而形成可涂抹的糊剂、凝胶、软膏剂、皂等,用于直接应用于使用者的皮肤。可以用于将化合物递送至皮肤的有用的皮肤用组合物的实例披露在美国专利US 4,608,392;美国专利US4,992,478;美国专利US 4,559,157;和美国专利US 4,820,508中。
可以通过比较体外活性和在动物模型中的体内活性确定本发明化合物和药物组合物的有用剂量。用于从小鼠和其它动物外推至人的有效剂量的方法是本领域公知的;例如参见美国专利US4,938,949。
本发明还涉及包含本发明化合物与药学上可接受的载体的药物组合物。适合于口服、局部或胃肠外给药的包含一定量化合物的药物组合物构成本发明的优选实施方案。在本发明上下文中对患者,特别是人的给药剂量应足以在合理的时间范围内在患者中获得治疗响应,而没有致命的毒性,且优选不产生超过可接受水平的副作用或发病。本领域技术人员公认剂量取决于各种因素,包括受试者的情况(健康),受试者的体重,同时治疗的类型(如果有的话),治疗频率,治疗剂的比例以及病情的严重性和阶段。
本发明还涉及用于抑制癌或肿瘤细胞或其中的Akt蛋白表达升高或具有组成型活性的其它细胞的存活或增殖或杀灭它们的方法,包括使所述细胞接触有效量的式I的化合物或其盐或类似物。在一个具体的实施方案中,该化合物具有式II所示的结构或其盐或类似物。在一个实施方案中,所述细胞是人细胞或其它哺乳动物细胞。可以使用本发明方法抑制或杀伤的癌细胞包括实质上是转移的那些细胞。因此,本发明还关注抑制癌或肿瘤细胞转移。可以在体外或体内实施所述方法。
本发明还涉及用于治疗人或动物肿瘤病症,诸如癌症或肿瘤的方法,包括对所述人或动物给予有效量的式I的化合物或其盐或其类似物。在一个具体的实施方案中,化合物具有式II中所示的结构或其盐或类似物。在一个实施方案中,对具有肿瘤病症及需要治疗肿瘤病症的患者给予有效量的本发明化合物或组合物。可以在化疗、免疫疗法和/或放疗之前、之后或同时给予所述抑制剂。本发明的方法可以任选包括鉴定需要或可能需要治疗肿瘤病症的患者。如果合适,可以使用医学或兽医专业人员公知的标准技术鉴定需要使用本发明方法治疗的患者。在一个实施方案中,所述患者可以是人或其它哺乳动物,诸如灵长类(猴子,黑猩猩,猿等),狗,猫,牛,猪或马或其它具有肿瘤病症的动物。本发明化合物对患者给药的给药途径和配制方法是本领域公知的,其实例如本文所述。本发明范围内的肿瘤病症包括,但不限于肛门、胆管、膀胱、骨、骨髓、肠(包括结肠和直肠)、乳腺、眼、胆囊、肾、口腔、喉、食道、胃、睾丸、宫颈、头、颈、卵巢、肺、间皮瘤、神经内分泌、阴茎、皮肤、脊髓、甲状腺、阴道、外阴、子宫、肝、肌肉、胰腺、前列腺、血细胞(包括淋巴细胞和其它免疫系统细胞)和脑的癌和/或肿瘤。本发明治疗的具体癌症包括癌、卡波西肉瘤、黑素瘤、间皮瘤软组织肉瘤、白血病(急性成淋巴细胞性白血病、急性髓样白血病、慢性淋巴细胞性白血病、慢性髓样白血病等)和淋巴瘤(何杰金瘤和非何杰金瘤)和多发性骨髓瘤。
为了治疗肿瘤病症,可以在使用其它抗肿瘤或抗癌药或物质(例如化疗剂、免疫治疗剂、放疗剂等)和/或放疗和/或手术治疗之前、之后或与之联合对有治疗需要的患者给予本发明的化合物和组合物,以除去肿瘤。例如,本发明的化合物和组合物可以用在治疗癌症的方法中,在该方法中将要使用或正在使用或已经使用以下治疗剂治疗了患者,所述治疗剂分别是:有丝分裂抑制剂,诸如泰素或长春碱,烷化剂,诸如环磷酰胺或异环磷酰胺,抗代谢药,诸如5-氟尿嘧啶或羟基脲,DNA嵌入剂,诸如阿霉素,或博来霉素,拓扑异构酶抑制剂,诸如依托泊苷或喜树碱,抗血管生成剂,诸如血管他丁,抗雌激素药,诸如他莫昔芬,和/或其它抗癌药或抗体,诸如,例如格列卫(诺华制药公司)和赫赛汀(Genentech,Inc.)。可以与本发明化合物同时或与本发明化合物相隔不同时间给予这些其它物质或放疗。本发明范围内的其它化疗剂的实例包括,但不限于六甲蜜胺,博来霉素,硼替佐米(VELCADE),白消安,亚叶酸钙,卡培他滨,卡铂,卡莫司汀,苯丁酸氮芥,顺铂,克拉屈滨,天冬酰胺酶(Crisantaspase),环磷酰胺,阿糖胞苷,达卡巴嗪,放线菌素D,柔红霉素,多西他赛,多柔比星,表柔比星,依托泊苷,氟达拉滨,氟尿嘧啶,吉非替尼(IRESSA),吉西他滨,羟基脲,伊达比星,异环磷酰胺,伊马替尼(GLEEVEC),伊立替康,多柔比星脂质体,洛莫司汀,美法仑,巯嘌呤,甲氨蝶呤,丝裂霉素,米托蒽醌,奥沙利铂,紫杉醇,喷司他丁,丙卡巴肼,雷替曲塞,链佐星,替加氟-尿嘧啶,替莫唑胺,塞替派,硫鸟嘌呤/硫鸟嘌呤,托泊替康,曲奥舒凡,长春碱,长春新碱,长春地辛,长春瑞滨。在一个典型的实施方案中,所述化疗剂是美法仑。本发明范围内的免疫治疗剂的实例包括,但不限于阿仑珠单抗,西妥昔单抗(ERBITUX),吉妥珠单抗,碘131托昔莫单抗,利妥昔单抗,曲妥珠单抗(HERCEPTIN)。本发明还涉及用于治疗肿瘤病症的方法,包括在给予化疗剂、免疫治疗剂、放疗剂或放疗之前、之后和/或同时给予有效量的本发明化合物。
本发明还涉及用于抑制细胞中Akt/PKB通路的方法,通过使所述细胞接触有效量的本发明化合物或组合物来进行。在一个实施方案中,所述化合物结合和抑制Akt1,AKT2和/或AKT3蛋白活性。在一个具体的实施方案中,所述化合物具有式II中所示的结构或其盐或类似物。在一个实施方案中,所述细胞是人或哺乳动物细胞,且可以是癌或肿瘤细胞或表现出异常的增殖、存活、迁移或分化的其它细胞。在一个实施方案中,所述细胞组成型表达或表达升高或异常水平的Akt蛋白,诸如Akt1,AKT2和/或AKT3。
本发明还涉及用于治疗具有与细胞中Akt蛋白组成型、异常或升高表达相关的病症的人或动物的方法,其中对所述人或动物给予治疗有效量的本发明化合物或组合物。所述病症的特征可以在于例如异常的细胞增殖、细胞存活、细胞迁移和/或细胞分化。在一个实施方案中,所述化合物结合和抑制Akt1,AKT2和/或AKT3蛋白活性。在一个具体的实施方案中,所述化合物具有式II所示的结构或其盐或类似物。
根据所治疗病症或疾病情况的不同,适当的剂量可以是减少靶细胞增殖或生长的用量。对于本文所述的癌症,适当的剂量是导致活性剂在癌组织诸如恶性肿瘤中的浓度(已知该浓度可实现所需的治疗响应)的剂量。优选的剂量是导致癌细胞生长得到最大抑制而无难以控制的副作用的用量。化合物的给药可以是连续的或在不同时间间隔,正如本领域技术人员可以确定的。
为了将这类剂量提供给所需的治疗,在某些实施方案中,以包含载体或稀释剂的组合物总重量为基准,本发明的药物组合物可以包含约0.1%至45%、且尤其是1至15%总重量的一种或多种化合物。例如,给予的活性成分的剂量水平可以是:静脉内,0.01至约20mg/kg;腹膜内,0.01至约100mg/kg;皮下,0.01至约100mg/kg;肌内,0.01至约100mg/kg;口服,0.01至约200mg/kg,且优选约1至100mg/kg;鼻内点滴,0.01至约20mg/kg;和气雾剂,0.01至约20mg/kg动物(体)重。
本发明还涉及试剂盒,其包含在一个或多个容器中的本发明的一种或多种化合物或包含本发明化合物或所述类似物或盐的组合物。在一个实施方案中,该试剂盒包含式II的化合物或其药学上可接受的盐或类似物。本发明的试剂盒可以任选包括药学上可接受的载体和/或稀释剂。在一个实施方案中,本发明的试剂盒包括一种或多种如本文所述的其它组分、助剂或佐剂。在另一个实施方案中,试剂盒包括一种或多种抗癌药,诸如本文所述的那些活性剂。在一个实施方案中,本发明的试剂盒包括描述如何给予试剂盒中的化合物或组合物的说明书或包装材料。试剂盒的容器可以是任何合适的材料,例如玻璃、塑料、金属等,并且具有任何适当的大小、性状或构造。在一个实施方案中,将本发明的化合物作为固体,诸如片剂、丸剂或粉末形式提供在试剂盒中。在另一个实施方案中,将本发明的化合物作为液体或溶液提供在试剂盒中。在一个实施方案中,该试剂盒包括含液体或溶液形式的本发明化合物的安瓿或注射器。
得益于本文所述治疗方法的哺乳动物种类包括,但不限于灵长类,诸如猿类,黑猩猩,猩猩,人,猴子;家养动物(例如宠物),诸如狗、猫、豚鼠、仓鼠、越南大肚猪、家兔和雪貂;驯化的农场动物,诸如牛、水牛、野牛、马、驴、猪、绵羊和山羊;一般在动物园可见的野生动物,诸如熊、狮、虎、豹、象、河马、犀牛、长颈鹿、羚羊、树懒、瞪羚、斑马、非洲大羚、场拨鼠、考拉熊、袋鼠、负鼠、浣熊、熊猫、土狼、海豹、海狮、海象、水獭、鼠海豚、海豚和鲸。可以得益于本文所述治疗方法的其它物种包括鱼、两栖动物、鸟类和爬行动物。本文所用的术语“患者”和“受试者”可以互换使用并且包括所述人和非人的种类。同样,本发明的体外方法可以对所述人和非人种类的细胞进行。
材料和方法
细胞系和NCI Diversity Set 本研究中使用的全部细胞系均购自ATCC或如上所述(Cheng等,1997;Jiang等,2000;Yang等,2004;Yang等,2005)。NCI Structural Diversity Set是选自约140,000-化合物NCI药物库的1,992种化合物的文库。在NCI DevelopmentalTherapeutics Program的网站上可以找到有关这些多样性组化合物的筛选、结构和活性的全面深入的数据(Jones等,1991)。
Akt-转化的细胞生长抑制的筛选.将AKT2转化的NIH3T3细胞或LXSN载体-转染的NIH3T3对照组细胞(Cheng等,1997)在96-孔组织培养培养板上铺板。在用5μM NCI Diversity Set化合物处理后,使用CellTier 96 One Solution Cell Proliferation试剂盒(Promega)检测细胞生长。将抑制AKT2-转化但非LXSN-转染的NIH3T3细胞的生长的化合物视为Akt抑制剂候选物并且进行进一步分析。
体外蛋白激酶,细胞存活和细胞凋亡测定.如上所述进行体外激酶测定(Jiang等,2000)。用MTT(Sigma)测定细胞存活。使用膜联蛋白V(BD Biosciences)检测细胞凋亡,根据制造商的说明书进行。重组Akt和PDK1购自Upstate Biotechnology Inc。
裸鼠肿瘤异种移植物模型中的抗肿瘤活性.收集肿瘤细胞,重新悬浮于PBS中并且如上所述皮下注入(s.c.)8周龄雌性裸鼠的右和左侧腹部(2x106个细胞/侧腹)(Sun等,1999)。当肿瘤达到约100-150mm3时,随机分组动物并且每日腹膜内注射(i.p.)媒介物或药物。对照组动物接受二甲亚砜(DMSO)(20%)媒介物,对受处理动物注射在20%DMSO中的API-1(10mg/kg/天)。
将本文涉及或引述的全部专利、专利申请、临时申请和公开文献完整地引入本文作为参考,包括所有的图和表,其引入程度使得它们与本说明书外在的教导一致。
下面是例证用于实施本发明方法的实施例。这些实施例不应被视为限制。除非另作陈述,否则本文所有百分比均按重量计且所有溶剂混合物比例均按体积计。
实施例1-通过筛选NCI Diversity Set对小分子Akt/PKB通路抑制剂-1,API-1的鉴定
由于Akt通路异常活化发生在几乎50%的所有人恶性肿瘤中且抑制Akt会诱导细胞生长停止和细胞凋亡的这一事实,从工业和学术界均对研发用于抗癌药物研发的小分子Akt抑制剂表现出了关注(Cheng等,2005;Granville等,2006)。尽管已经报导了12种Akt抑制剂,但是它们中的许多缺乏体内抗肿瘤活性。已经报导了基于脂质的Akt抑制剂哌立福辛处于I和II期研究中(Van Ummersen等,2004;Bailey等,2006)。然而,在这些研究中无一评价了Akt的调控作用。近期哌立福辛在胰腺癌中的II期研究因在第一阶段过程中发生不可接受的不良反应而被终止(Marsh等,2007)。然而,对研发可避免抑制其它激酶活性且不良反应最低的有效选择性Akt抑制剂存在需求。为了鉴定Akt的小分子抑制剂,我们如“材料和方法”中所述评价了来自NCI(美国国家癌症研究所)的能够抑制AKT2-转化的、但并非空载体LXSN-转染的NIH3T3细胞生长的活性剂的1,992个化合物的化学文库(NCI Diversity Set)。三次实验表明32种化合物仅抑制AKT2-转化细胞的生长。我们预先表征了其中之一,即API-2/曲西立滨,它是具有体外抗肿瘤活性且目前处于I期临床试验中的全-Akt抑制剂(Yang等,2004)。
在目前的研究中我们证实了Akt/PKB抑制剂-1(API-1)特异性抑制活细胞中Akt的激酶活性和磷酸化水平。图1A表示API-1的化学结构(NSC 177223),它没有化学名称并且尚未在NCI 60细胞系中测试(http://dtp.nci.nih.gov/)。由于API-1选择性抑制AKT2转化的细胞超过了未转化的亲代细胞,所以我们首先检验了API-1是否为AKT2激酶抑制剂且它是否还抑制Akt的其它两个成员。用HA-标记的野生型Akt1、AKT2和AKT3转染HEK293细胞。在血清饥饿过夜后,用API-1将细胞处理60min,然后进行EGF刺激并且用抗-HA抗体免疫沉淀。对免疫沉淀物进行体外激酶测定。图1B表示API-1抑制胰岛素-诱导的Akt1、AKT2和AKT3的激酶活性。我们接下来检验了API-1是否抑制活细胞中的Akt。用不同剂量的API-1将表达升高水平的磷酸-Akt的OVCAR3细胞处理3小时。用抗-磷酸-Akt-S473抗体进行的免疫印迹分析表明API-1有效降低Akt磷酸化水平且IC50为约0.8M。然而,总Akt水平未改变(图1C)。此外,我们检验了API-1是否直接抑制体外Akt激酶活性。将重组组成型活性Akt蛋白与Akt/SGK底物肽(Upstate)一起在含不同量API-1和化合物E,即全(pan)-激酶ATP-竞争者抑制剂(包括Akt)的激酶缓冲液中孵育作为阳性对照。三次实验未显示API-1对Akt激酶活性的作用(图1D),从而提示API-1不直接抑制体外Akt并且API-1既不作为ATP、也不作为底物竞争者起作用。
实施例2-API-1确实不抑制Akt的上游激活物
Akt被细胞外刺激物和细胞内信号分子通过被PTEN负调节的PI3K-依赖性方式活化。PI3K的活化或PTEN突变会活化PDK1,导致诱导Akt激酶活性。因此,API-1抑制Akt可能因靶向Akt的上游分子,诸如PI3K和PDK1而导致。为了这一目的,我们接下来检验了API-1是否抑制PI3K和/或PDK1。使HEK293细胞血清饥饿,然后在EGF刺激前用API-1或PI3K抑制剂渥曼青霉素处理1小时。用抗-p110α抗体免疫沉淀PI3K。使用PI-4-P作为底物对免疫沉淀物进行体外PI3K激酶测定。正如图2A中所示,EGF-诱导的PI3K活性被渥曼青霉素而非API-1抑制。为了评价API-1对PDK1的作用,我们在体外使用SGK激酶作为读出值进行了PDK1激酶测定(Upstate BiotechnologyInc.)。不同于PDK1抑制剂UCN-01(Sato等,2002),API-1在体外对PDK1激酶活性无影响(图2B)。为了进一步评价API-1对活细胞中PDK1活化的作用,我们在API-1处理OVCAR3细胞后检验了PDK1-Ser241(即,自磷酸化的且对其活性而言至关重要的残余物)的磷酸化水平(Casamayor等,1999)。图2D表示PDK1磷酸化水平不被API-1抑制。
实施例3-API-1对Akt的高度选择性超过了对AGC激酶成员PKA、PKC和SGK以及其它信号传导分子ERK,JNK,p38和STAT3
Akt属于AGC(PKA/PKG/PKC)激酶家族,该家族还包括PKA,PKC,血清-和糖皮质激素-诱导型激酶(SGK),p90核糖体S6激酶,p70S6K,促分裂原-和应激-活化蛋白激酶和PKC-相关激酶。在AGC激酶家族中,PKA,PKC和SGK的蛋白结构与Akt激酶远比其它成员接近。因此,我们接下来检验了API-1对这3种激酶酶活性的作用。用预孵育的所示剂量的API-1或特异性抑制剂与重组PKA或SGK蛋白进行体外PKA激酶测定和SGK激酶测定30分钟,然后通过在激酶缓冲液中添加肯普肽或Akt/SGK底物肽和[γ-32P]ATP来启动激酶测定。体外激酶测定表明PKA和SGK的激酶活性分别被PKAI和化合物E抑制,而API-1未表现出对它们的活性的作用(图2C和2E)。为了进一步评价API-1对活细胞中PKA和PKCα活化的作用,用所示剂量的API-1或PKA和PKC的特异性抑制剂处理OVCAR3细胞,免疫印迹分析表明PKA和PKC的磷酸化水平不被API-1抑制(图2D)。此外,用HA-标记的SGK转染HEK293细胞。体外激酶测定表明EGF-诱导的SGK激酶活性被渥曼青霉素、而非API-1减弱(图2F)。
为了确定API-1是否对其它癌基因存活途径有影响,用API-1(10αM)将OVCAR3细胞处理不同次数并且用商购的抗-磷酸-抗体进行免疫印迹。我们未观察到在用API-1处理后可检测到的Stat3,JNK,p38和Erk1/2的磷酸化水平改变(图2G)。这些数据表明API-1可以特异性抑制Akt通路。
实施例4-API-1抑制组成型活性Akt及其下游靶标
由于API-1不能在体外直接抑制Akt但能在活细胞中消除Akt的激酶活性和磷酸化,而对PI3K和PDK1无影响,所以这导致我们推定该化合物可以与Akt蛋白发生相互作用,但不与ATP-结合位点发生相互作用,从而防止了Thr308和Ser473被PDK1和PDK2磷酸化。如果这是可靠的,那么API-1还应抑制组成型活性Akt的活化,因为其活化仍然需要Thr308和Ser473的磷酸化(Sun等,2001)。为了测试这一推断,用HA-标记的Myr-Akt1、Myr-Akt2和Myr-Akt1E40K转染HEK293细胞。在血清饥饿过夜后,用或不用API-1处理细胞。使用抗-HA抗体免疫沉淀Myr-Akt1,Myr-Akt2和Akt1-E40K。对免疫沉淀物进行体外激酶测定并且使用抗-磷酸-Akt-T308抗体进行免疫印迹分析。图3A表示Myr-Akt1,Myr-Akt2和Akt1-E40K的体外Akt激酶活性和磷酸化水平被API-1抑制,从而支持了API-1可以结合Akt分子以防止其在细胞内活化的论断。
Akt通过大量蛋白质的磷酸化发挥其细胞功能(Datta等,1999)。我们接下来检验了API-1是否抑制Akt的下游靶标。由于GSK3β和mTOR是两种主要的Akt靶标,所以我们评价了API-1对GSKβ和S6(即p70S6K底物)的磷酸化水平的作用。用API-1处理OVCAR3后,免疫印迹分析显示API-1极大地抑制了它们的磷酸化(图3B)。
实施例5-API-1抑制Akt-超表达/活化的人癌细胞系中的细胞生长并且诱导细胞凋亡
Akt是主要的前-生长和前-存活途径。具有升高的Akt激酶的癌细胞表现出对化疗药诱导的细胞生长停止和细胞死亡更大的抗性,而减少Akt对不同凋亡前刺激物诱导的细胞凋亡的敏感度(Solit等,2003;Xu等,2003;Jetzt等,2003)。API-1选择性抑制Akt通路的能力提示它应该能在那些具有异常Akt表达/活化的肿瘤细胞中抑制增殖和/或优先诱导细胞凋亡。为了测试这一判断,API-1用于处理因Akt超表达(OVCAR3,OVCAR8,MCF7和PANC1)或PTEN基因突变(MDA-MB-468PC-3和LNCaP)而产生的表达组成型活性Akt的细胞和不表达组成型活性Akt的细胞(OVCAR5,MDA-MB-435s,DU-145和COLO357)。免疫印迹分析表明API-1在表达升高的Akt的细胞中显著抑制Akt磷酸化水平,而在表现出低水平Akt的细胞系中API-1也使磷酸-Akt减少(图4A且数据未显示)。但是,API-1诱导PARP裂解和抑制细胞生长的程度在Akt-超表达/活化的细胞中远高于在具有低水平Akt的那些细胞中(图4A,4B和4C)。API-1处理抑制细胞增殖在Akt-超表达/活化的细胞系OVCAR3,OVCA8,MDA-MB-468和MCF7中约为50-70%,而在OVCAR5和MDA-MB-435s细胞中仅约为10-30%(图4B和4C)。此外,API-1在OVACAR3和MDA-MB-468中诱导细胞凋亡分别为9-倍和4.6-倍,而在API-1-处理的OVCAR5和MDA-MB-435s细胞中观察到很少的细胞凋亡(图4D-4G)。因此,API-1抑制细胞生长并且优先在表达异常Akt的细胞中诱导细胞凋亡。
实施例6-API-1抑制Akt超表达的裸鼠中的肿瘤生长
预先证实通常在人卵巢和胰腺癌中检测到Akt异常活化和超表达(Cheng等,1992),且Akt的反义物显著抑制肿瘤细胞生长和侵入(Cheng等,1996)。此外,PI3K,HSP70,Src和法尼基转移酶的抑制剂抑制Akt通路导致细胞生长停止和诱导细胞凋亡(Solit等,2003;Xu等,2003)。因为API-1抑制Akt信号传导并且诱导具有升高水平Akt的癌细胞中的细胞凋亡和细胞生长停止(图4A-4G),所以理由充分的是在裸鼠中具有升高水平Akt的肿瘤生长对API-1应比具有低水平Akt的肿瘤生长对API-1更敏感。就这一目的而言,将超表达Akt的细胞(OVCAR3和PANC-1)s.c.(皮下)植入左侧腹,并且将表达低水平Akt的那些细胞系(OVCAR5和COLO357)s.c.植入小鼠右侧腹。当肿瘤达到约100-150mm3的平均大小时,对动物随机分组并且用媒介物或API-1进行i.p.(静脉注射)处理(10mg/kg/天)。正如图5A-5J中例证的,用媒介物对照品处理的OVCAR3和PANC1肿瘤持续生长。API-1抑制OVCAR3和PANC1肿瘤生长分别为70%和50%(图5C-5F和5G-5J)。相反,API-1对裸鼠中OVCAR5和COLO357细胞生长几乎无作用(图5A-5J)。在10mg/kg/天的剂量下,API-1对小鼠的血糖水平、体重、活动和摄食无影响(数据未显示)。在处理的肿瘤样品中,Akt的磷酸化水平被API-1降低了约70%,而总Akt含量无改变(图5K-5L)。这些结果共同表明API-1选择性抑制具有升高水平的Ak t的肿瘤生长。
应理解本文所述的实施例和实施方案仅用于例证目的,并且提示本领域技术人员根据它们进行各种变型或改变,且这些变型或改变包括在本申请精神和范围以及待批权利要求范围内。此外,本文披露的任意本发明的任意要素或限制或其实施方案可以与本文披露的任意和/或所有其它要素或限制(单独或以任意组合方式)或任意其它发明或其实施方案组合,且所有这些组合均属于本发明范围内,但不限于此。
具有抗肿瘤活性的AKT/PKB抑制剂专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。


![酞嗪并[1,2,b]喹唑啉-8-酮化合物及其制备方法和在抗肿瘤药物中的应用](https://www.zhichawang.com/images/tongyong/wutu.jpg)







![一种β-(2,3-二氢-萘[1,2,e]-m-噁嗪)并四苯基卟啉化合物及其制备方法和应用](https://www.zhichawang.com/images/CN106366086A/CN106366086A.jpg)
![苯并[e][1,2,4]三嗪-1-氧衍生物及其组合物和应用](https://www.zhichawang.com/images/CN106831711A/CN106831711A.jpg)

动态评分
0.0