








专利摘要
本发明涉及一种埃罗替尼中间体的制备方法,具体包括如下步骤:将化合物2加入氨水中,室温下加入催化量的四丁基溴化铵,超声5‑30min,生成化合物3;超声频率为30‑50kHz。
权利要求
1.一种制备中间体化合物3的方法,其特征在于包括如下步骤:
(1)
将化合物1溶于有机溶剂中,室温下加入多聚甲醛和催化量的三氟化硼乙醚,加热50-60℃反应3-5h,生成化合物2;化合物1与多聚甲醛的摩尔比为1:10-20;有机溶剂选自DMF、DMA或THF;
(2)
将化合物2加入氨水中,室温下加入催化量的四丁基溴化铵,超声5-30min,生成化合物3;超声频率为30-50kHz。
2.权利要求1所述的方法在制备埃罗替尼中的应用。
说明书
技术领域
本发明属于药物合成领域,具体涉及一种埃罗替尼中间体的制备方法。
背景技术
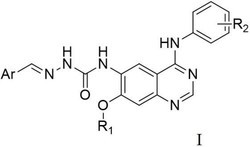
盐酸埃罗替尼(Erlotinib Hydrochloride)是美国OSI Pharmaceuticals公司开发的一种酪氨酸酶抑制剂,目前临床主要用于非小细胞肺癌的治疗。其化学名为:N-(3-乙炔苯基)-[6,7-二(2-甲氧基乙氧基)]喹唑啉-4-胺盐酸盐。化学结构如下所示:
埃罗替尼的合成路线主要是美国辉瑞公司报道的美国专利US5747498A,其合成路线如下:
上述合成路线中,erlo-4的合成需加热至160℃左右,且反应收率不高,因此开发一种合成erlo-4的新方法用于合成埃罗替尼成为研究的重点。
发明内容
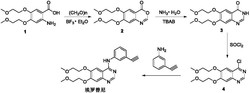
本发明提供一种合成埃罗替尼的方法,其特征在于包括如下步骤:
(1)化合物1与多聚甲醛在催化量三氟化硼乙醚的条件下反应生成化合物2;
(2)化合物2与氨水在催化量四丁基溴化铵及超声条件下反应生成化合物3;
(3)化合物3与氯化亚砜反应生成化合物4;
(4)化合物4与间氨基苯乙炔反应生成埃罗替尼。
上述合成埃罗替尼的方法,其特征在于步骤(1)中化合物1与多聚甲醛的摩尔比为1:10-20,反应温度为50-60℃;步骤(2)中超声反应时间为5-30min,反应温度为室温,超声频率为30-50kHz;步骤(3)反应温度为60-80℃;步骤(4)反应温度为70℃至回流温度,溶剂为异丙醇;上述步骤(1)、(3)中任选加入有机溶剂,所述有机溶剂优选DMF、DMA或THF。
本发明的另一实施方案中提供一种用于合成埃罗替尼的中间体,其特征在于所述中间体具有化合物2所示结构:
本发明的另一实施方案提供中间体化合物2在制备埃罗替尼中的用途。
本发明的另一实施方案提供中间体化合物2在制备化合物3中的用途。
本发明的另一实施方案提供一种制备中间体化合物2的方法,其特征在于包括如下步骤:
化合物1溶于有机溶剂中,室温下加入多聚甲醛和催化量的三氟化硼乙醚,加热50-60℃反应3-5h,生成化合物2;有机溶剂优选DMF、DMA或THF。
本发明的另一实施方案提供一种制备中间体化合物3的方法,其特征在于包括如下步骤:
将化合物2加入氨水中,室温下加入催化量的四丁基溴化铵,超声5-30min,生成化合物3;超声频率为30-50kHz。
上述制备中间体化合物3的方法,还包括如下步骤:
将化合物1溶于有机溶剂中,室温下加入多聚甲醛和催化量的三氟化硼乙醚,加热50-60℃反应3-5h,生成化合物2;有机溶剂优选DMF、DMA或THF。
与现有技术相比本发明的优点在于:
(1)本发明提供一种制备埃罗替尼的新中间体即化合物2,该中间体可由化合物1在多聚甲醛、三氟化硼乙醚作用下,于50-60℃即可反应生成中间体化合物,所用试剂廉价易得,反应条件温和,转化率达99%;
(2)本发明由化合物2制备化合物3的反应条件温和,反应时间短,转化率高。
具体实施方式
为了便于对本发明的进一步理解,下面提供的实施例对其做了更详细的说明。但是这些实施例仅供更好的理解发明而并非用来限定本发明的范围或实施原则,本发明的实施方式不限于以下内容。
实施例1
称取化合物1(285mg,1.0mmol)溶于DMF(10mL)中,室温下加入多聚甲醛(300mg,10mmol)和催化量的三氟化硼乙醚,加热至50℃,搅拌反应,TLC检测反应原料(化合物1)完全消失(约反应5h),停止加热降至室温,将反应液倒入冰水中(约100mL),搅拌5-10min,抽滤得滤饼,滤饼加入20mL乙腈,加热50℃至固体完全溶解,自然降至室温结晶,抽滤,滤饼用乙腈洗2次,真空干燥得白色固体292mg(收率约为98.9%),即为化合物2(ESI-MS(m/z):296.11[M+H]+,318.09[M+Na]+,1H NMR(400MHz,CDCl3):δ8.23(s,1H,H-2),7.50(s,1H,H-5),7.18(s,1H,H-8),4.28-4.20(m,8H,CH2×4),3.43(s,3H,CH3O),3.40(s,3H,CH3O))。
实施例2
称取化合物1(285mg,1.0mmol)溶于THF(10mL)中,室温下加入多聚甲醛(600mg,20mmol)和催化量的三氟化硼乙醚,加热至60℃,搅拌反应,TLC检测反应原料(化合物1)完全消失(约反应3h),停止加热降至室温,将反应液倒入冰水中(约100mL),搅拌5-10min,抽滤得滤饼,滤饼加入20mL乙腈,加热50℃至固体完全溶解,自然降至室温结晶,抽滤,滤饼用乙腈洗2次,真空干燥得白色固体293mg(收率约为99.2%),即为化合物2(结构确证数据与实施例1一致)。
实施例3
称取化合物1(285mg,1.0mmol)溶于DMF(10mL)中,室温下加入多聚甲醛(300mg,10mmol),加热至50℃,搅拌反应(约反应5h),TLC检测显示反应液中化合物1仍为主点,即在不加三氟化硼乙醚的情况下,几乎反应。
实施例4
称取化合物1(285mg,1.0mmol)溶于原甲酸三乙酯(10mL)中,室温下加入催化量的对甲苯磺酸,加热至回流温度反应4h后,停止加热降至室温,将反应液倒入冰水中(约100mL),搅拌5-10min,抽滤得滤饼,滤饼加入20mL乙腈,加热50℃至固体完全溶解,自然降至室温结晶,抽滤,滤饼用乙腈洗1-2次,真空干燥得白色固体186mg(收率约为63.0%),即为化合物2(结构确证数据与实施例1一致)。
实施例5
称取化合物2(295mg,1.0mmol)置于氨水(10mL)中,室温下加入催化量的四丁基溴化铵,超声反应5min,超声频率为50kHz,TLC检测反应原料(化合物2)完全消失,用二氯甲烷稀释(30mL),依次用水、饱和NaCl洗涤,有机相浓缩后,加入15mL乙腈,加热50℃至固体完全溶解,自然降至室温结晶,抽滤,滤饼用乙腈洗2次,真空干燥得白色固体292mg(收率约为99.2%),即为化合物3(熔点:182℃,ESI-MS及1H NMR与文献European Journal of Medicinal Chemistry,Volume 43,Issue 7,July 2008,Pages 1478–1488报道一致)。
实施例6
称取化合物2(295mg,1.0mmol)置于氨水(10mL)中,室温下加入催化量的四丁基溴化铵,超声反应30min,超声频率为30kHz,TLC检测反应原料(化合物2)完全消失,用二氯甲烷稀释(30mL),依次用水、饱和NaCl洗涤,有机相浓缩后,加入15mL乙腈,加热50℃至固体完全溶解,自然降至室温结晶,抽滤,滤饼用乙腈洗2次,真空干燥得白色固体291mg(收率约为98.9%),即为化合物3(结构确证数据与实施例5一致)。
实施例7
称取化合物3(294mg,1.0mmol)溶于DMF(1mL)中,室温下加入氯化亚砜(2mL),加热至80℃,反应4-5小时,TLC检测反应原料(化合物3)完全消失,停止加热,蒸除氯化亚砜,加入少量石油醚(1mL)出现大量固体,蒸除石油醚,用二氯甲烷稀释(40mL),依次用饱和NaHCO3、饱和NaCl洗涤,有机相用无水硫酸钠干燥,过滤,浓缩、干燥得微黄色固体(295mg,收率约为94.3%)即为化合物4(HPLC检测纯度为99.1%,1H NMR(DMSO-d6):δ8.88(s,1H),7.50(s,1H),7.46(s,1H),4.38-4.35(m,4H),3.78-3.75(m,4H),3.37-3.34(m,6H))。
实施例8
称取化合物3(589mg,2.0mmol)溶于DMF(2mL)中,室温下加入氯化亚砜(4mL),加热至60℃,反应4-5小时,TLC检测反应原料(化合物3)完全消失,停止加热,蒸除氯化亚砜,加入少量石油醚(1mL)出现大量固体,蒸除石油醚,用二氯甲烷稀释(60mL),依次用饱和NaHCO3、饱和NaCl洗涤,有机相用无水硫酸钠干燥,过滤,浓缩、干燥得微黄色固体(598mg,收率约为95.6%)即为化合物4(HPLC检测纯度为99.5%,结构确证数据与实施例7一致)。
实施例9
称取化合物4(313mg,1.0mmol)加入异丙醇(5mL)中,室温下加入间氨基苯乙炔(200mg),加热至回流温度(升温过程中溶液逐渐澄清,稍后有大量白色固体出现),反应3-4小时,TLC检测反应原料(化合物4)完全消失,停止加热,降至室温,抽滤,滤饼用热的异丙醇洗涤2次,真空干燥得白色固体(395mg),经正丁醇重结晶得埃罗替尼纯品(380mg,收率约为96.6%,1H NMR与现有技术一致,HPLC检测纯度为99.5%)。
实施例10
称取化合物4(626mg,2.0mmol)加入异丙醇(10mL)中,室温下加入间氨基苯乙炔(400mg),加热至70℃(升温过程中溶液逐渐澄清,稍后有大量白色固体出现),反应3-4小时,TLC检测反应原料(化合物4)完全消失,停止加热,降至室温,抽滤,滤饼用热的异丙醇洗涤2次,真空干燥得白色固体(792mg),经正丁醇重结晶得埃罗替尼纯品(750mg,收率约为95.3%,HPLC检测纯度为99.7%)。
一种埃罗替尼中间体的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











动态评分
0.0