





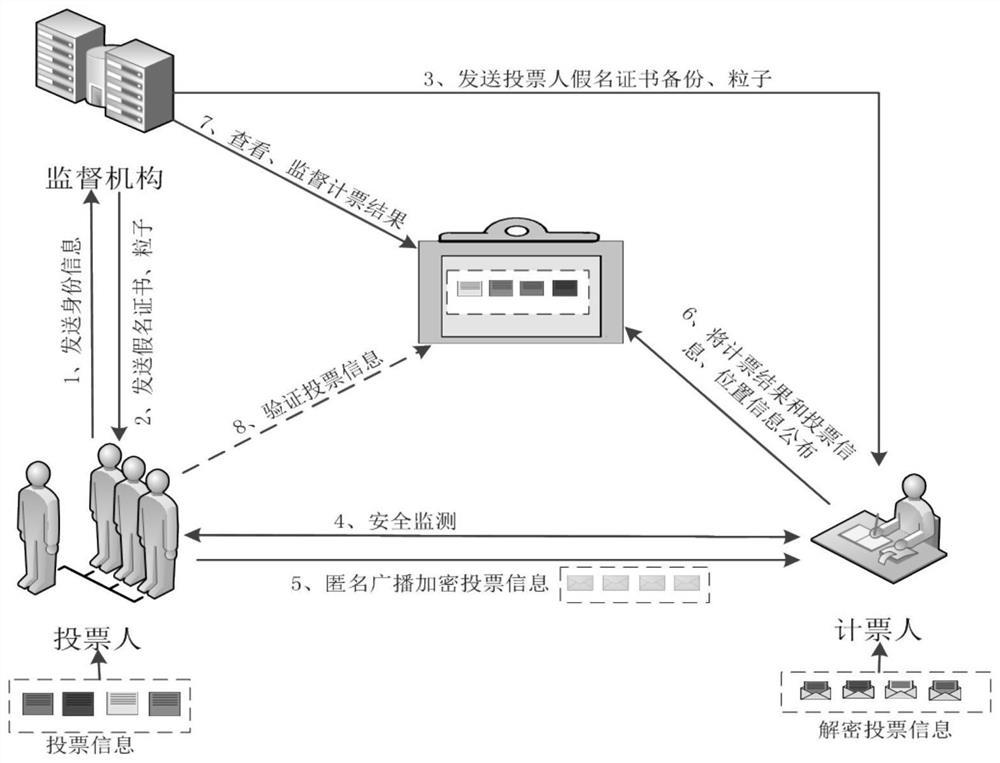

专利摘要
本发明公开了一种黄连素重要中间体的合成。在有机溶剂中,胡椒环与氯气反应得5‑氯代胡椒环;在醚类溶剂下,5‑氯代胡椒环与镁条反应,得格氏试剂;在有机溶剂中,2,3‑二甲氧基苯甲酸在脱水剂的作用下与氮杂环丙烷反应得1‑氮丙啶‑2,3‑二甲氧基苯基乙酮;在格氏试剂的作用下,1‑氮丙啶‑2,3‑二甲氧基苯基乙酮经开环得黄连素重要中间体N‑(2‑(苯并[d][1,3]二氧杂环戊烯‑5‑基)乙基)‑2,3‑二甲氧基苯甲酰胺。本发明反应条件温和,操作简单。
权利要求
1.一种黄连素重要中间体的合成,其特征在于,包括以下步骤:
(1)在有机溶剂中,胡椒环与氯气反应得5-氯代胡椒环,所述的有机溶剂为氯仿、二氯甲烷、稀盐酸、二硫化碳、四氯化碳的一种,反应温度为20℃~80℃;
(2)在醚类溶剂下,5-氯代胡椒环在碘单质或1,2-溴乙烷的作用下与镁条反应,制得格氏试剂。所述的醚类溶剂为四氢呋喃、甲基四氢呋喃、乙醚、1,4-二氧六环的一种,反应温度为10℃~68℃;
(3)在有机溶剂中,2,3-二甲氧基苯甲酸在脱水剂的作用下与氮杂环丙烷反应得1-氮丙啶-2,3-二甲氧基苯基乙酮,所述的有机溶剂为二氯甲烷、甲苯、二甲苯、N,N-二甲基甲酰胺的一种,其中脱水剂为N,N-二环己基碳二亚胺(DCC)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)、多聚磷酸、五氧化二磷、三氯氧磷的其中一种,反应温度为0℃~100℃;
(4)在有机溶剂中,1-氮丙啶-2,3-二甲氧基苯基乙酮与格氏试剂亲核开环,制得黄连素重要中间体N-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)-2,3-二甲氧基苯甲酰胺,所述的有机溶剂为四氢呋喃、乙醚、甲基四氢呋喃、1,4二氧六环的一种,催化剂为CuBr、CuI、CuBr.SMe2中任一种,反应温度为-40℃~68℃。
2.如权利要求书1所述的黄连素重要中间体N-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)-2,3-二甲氧基苯甲酰胺的合成,其特征在于,步骤(1)中,所述的有机溶剂是氯仿,反应温度为35℃。
3.如权利要求书1所述的黄连素重要中间体N-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)-2,3-二甲氧基苯甲酰胺的合成,其特征在于,步骤(2)中,所述的有机溶剂是四氢呋喃,反应温度为30℃。
4.如权利要求书1所述的黄连素重要中间体N-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)-2,3-二甲氧基苯甲酰胺的合成,其特征在于,步骤(3)中,所述的有机溶剂是二氯甲烷,脱水剂为1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI),反应温度为25℃。
5.如权利要求书1所述的黄连素重要中间体N-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)-2,3-二甲氧基苯甲酰胺的合成,其特征在于,步骤(4)中,所述的有机溶剂是四氢呋喃,催化剂为CuBr.SMe2,反应温度为0℃。
说明书
技术领域
本发明属于药物化学技术领域,涉及一种盐酸黄连素关键中间体,即N-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)-2,3-二甲氧基苯甲酰胺的合成。
技术背景
黄连素是一种重要的异喹啉类生物碱,可从黄连、黄柏、三颗针、十大功劳等植物的根茎中提取。它具有广谱抗菌作用,临床上可用于治疗呼吸道、肠炎、菌痢、妇科炎症以及泌尿道感染。近年来随着研究的不断深入,发现黄连素有增强心肌收缩力,降低外周血管阻力,明显改善心功能的作用。此外,黄连素还有明显的抗心律失常作用,且无明显的致心律失常等副作用。
在已有的黄连素合成路线中,大多数采用胡椒乙胺作为关键的中间体,以N-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)-2,3-二甲氧基苯甲酰胺为黄连素关键中间体合成黄连素研究较少,但以它作为黄连素重要中间体的合成是非常有价值的。Decker等报道了以胡椒醛为起始原料,经过硝化、还原、霍夫曼重排合成黄连素重要中间体,该路线需催化氢化,给操作者带来不便;Gatland等和Reddy等报道了通过有机钯催化剂合成黄连素,尽管这些合成路线的收率较高,但都存在一些不足,如原料不易得、使用昂贵有机钯催化剂、工业化成本太高等:东北制药采用苯酚为原料,经胡椒醛、Darzen反应得黄连素重要中间体,但该方法路线较长,收率较低。基于这些路线的不足,需寻找合成盐酸黄连素重要中间体的新方法。
发明内容
本发明的目的在于克服上述技术存在的不足,提供一种收率高、操作简单、成本低的黄连素重要中间体的合成。
本发明具体技术方案为:
一种黄连素重要中间体的合成,包括以下步骤:
(1)在有机溶剂中,胡椒环与氯气反应得5-氯代胡椒环,所述的有机溶剂为氯仿、二氯甲烷、稀盐酸、二硫化碳、四氯化碳的一种,反应温度为20℃~80℃;
(2)在醚类溶剂下,5-氯代胡椒环在碘单质或1,2-溴乙烷的作用下与镁条反应,制得格氏试剂。所述的醚类溶剂为四氢呋喃、甲基四氢呋喃、乙醚、1,4-二氧六环的一种,反应温度为10℃~68℃;
(3)在有机溶剂中,2,3-二甲氧基苯甲酸在脱水剂的作用下与氮杂环丙烷反应得1-氮丙啶-2,3-二甲氧基苯基乙酮,所述的有机溶剂为二氯甲烷、甲苯、二甲苯、N,N-二甲基甲酰胺的一种,其中脱水剂为N,N-二环己基碳二亚胺(DCC)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)、多聚磷酸、五氧化二磷、三氯氧磷的其中一种,反应温度为0℃~100℃;
(4)在有机溶剂中,1-氮丙啶-2,3-二甲氧基苯基乙酮与格氏试剂亲核开环,制得黄连素重要中间体N-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)-2,3-二甲氧基苯甲酰胺,所述的有机溶剂为四氢呋喃、乙醚、甲基四氢呋喃、1,4-二氧六环的一种,催化剂为CuBr、CuI、CuBr.SMe2中任一种,反应温度为-40℃~68℃。
优选地,步骤(1)中,所述有机溶剂为氯仿,反应温度为35℃。
优选地,步骤(2)中,所述有机溶剂为四氢呋喃,反应温度为30℃。
优选地,步骤(3)中,所述有机溶剂为二氯甲烷,脱水剂为1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI),反应温度为25℃。
优选地,步骤(4)中,所述有机溶剂为四氢呋喃,催化剂为CuBr.SMe2,反应温度为0℃。
与现有技术相比,本发明的有益效果为:
本发明是一种收率高、操作简单、成本低的黄连素重要中间体的合成,该方法反应条件温和、原子经济性好。
附图说明
图1是本发明黄连素重要中间体的合成的路线图。
具体实施方式
下面结合具体附图和实施例对本发明的技术方案作进一步详细地说明。
参照图1,本发明黄连素重要中间体的合成,即N-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)-2,3-二甲氧基苯甲酰胺的合成。在有机溶剂中,胡椒环与氯气反应得5-氯代胡椒环;在醚类溶剂下,5-氯代胡椒环与镁条反应,得格氏试剂;在有机溶剂中,2,3-二甲氧基苯甲酸在脱水剂的作用下与氮杂环丙烷反应得1-氮丙啶-2,3-二甲氧基苯基乙酮;在格氏试剂的作用下,1-氮丙啶-2,3-二甲氧基苯基乙酮经开环得N-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)-2,3-二甲氧基苯甲酰胺。
步骤一:5-氯代胡椒环的制备
实施例1:于装有搅拌器、回流冷凝器,滴液漏斗的反应瓶中,加入0.5000g胡椒环,10mL氯仿,搅拌溶解。于滴液漏斗中加入5mL氯仿,慢慢通入1.005g氯气,控制反应液温度35℃,滴加氯气的氯仿溶液,约5min加完。加完后继续搅拌反应2h,然后用水洗两次,合并有机层,饱和食盐水洗涤,经无水硫酸钠干燥,蒸出氯仿后得产物,收率为96.2%,1H NMR(400MHz,CDCl3)δ=6.75-6.81(m,2H,HAr),6.69(dd,1H,HAr),5.94(s,2H,CH2)。
实施例2:于装有搅拌器、回流冷凝器,滴液漏斗的反应瓶中,加入0.5000g胡椒环,10mL氯仿,搅拌溶解。于滴液漏斗中加入5mL氯仿,慢慢通入1.005g氯气,控制反应液温度60℃,滴加氯气的氯仿溶液,约5min加完。加完后继续搅拌反应2h,然后用水洗两次,合并有机层,饱和食盐水洗涤,经无水硫酸钠干燥,蒸出氯仿后得产物,收率为94.1%,1H NMR(400MHz,CDCl3)δ=6.75-6.81(m,2H,HAr),6.69(dd,1H,HAr),5.94(s,2H,CH2)。
实施例3:
于装有搅拌器、回流冷凝器,滴液漏斗的反应瓶中,加入0.5000g胡椒环,10mL氯仿,搅拌溶解。于滴液漏斗中加入5mL氯仿,慢慢通入1.005g氯气,控制反应液温度80℃,滴加氯气的氯仿溶液,约5min加完。加完后继续搅拌反应2h,然后用水洗两次,合并有机层,饱和食盐水洗涤,经无水硫酸钠干燥,蒸出氯仿后得产物,收率为90.2%,1H NMR(400MHz,CDCl3)δ=6.75-6.81(m,2H,HAr),6.69(dd,1H,HAr),5.94(s,2H,CH2)。
步骤二:格氏试剂的制备
实施例4:
将0.1352g镁条、0.0500g碘依次加入干燥的三颈瓶,N2保护,再加入4mL干燥的四氢呋喃,滴加一部分5-氯代胡椒环引发,待引发成功后,缓慢滴加剩下的5-氯代胡椒环,共加入0.8013g 5-氯代胡椒环。30℃反应0.5h,以备下一步反应。
实施例5:
将0.1352g镁条、0.0500g碘依次加入干燥的三颈瓶,N2保护,再加入4mL干燥的四氢呋喃,滴加一部分5-氯代胡椒环引发,待引发成功后,缓慢滴加剩下的5-氯代胡椒环,共加入0.8013g 5-氯代胡椒环。60℃反应0.5h,以备下一步反应。
实施例6:
将0.1352g镁条、0.0500g碘依次加入干燥的三颈瓶,N2保护,再加入4mL干燥的甲基四氢呋喃,滴加一部分5-氯代胡椒环引发,待引发成功后,缓慢滴加剩下的5-氯代胡椒环,共加入0.8013g 5-氯代胡椒环。60℃反应0.5h,以备下一步反应。
步骤三:1-氮丙啶-2,3-二甲氧基苯基乙酮的制备
实施例7:
将0.5000g 2,3-二甲氧基苯甲酸、0.7750gl-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)和8mL干燥的二氯甲烷依次加入干燥的三颈瓶中。然后降温至0℃,将0.1125g氮杂环丙烷溶于7.5mL二氯甲烷所配成的混合溶液滴加到反应液中,大约滴加3min。滴毕,在0℃反应2h后停止反应。用水洗两次,合并有机层,饱和食盐水洗涤,经无水硫酸钠干燥,蒸出二氯甲烷后经柱层析分离得产物,收率为93.0%。1H NMR(400MHz,CDCl3)δ=7.33-7.35(dd,1H,Ar),7.02-7.11(m,2H,Ar),3.88(s,3H,OCH3),3.86(s,3H,OCH3),2,.35(s,4H,CH2)ppm。
实施例8:
将0.5000g 2,3-二甲氧基苯甲酸、0.7750g1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)和8mL干燥的二氯甲烷依次加入干燥的三颈瓶中。然后降温至0℃,将0.1125g氮杂环丙烷溶于7.5mL二氯甲烷所配成的混合溶液滴加到反应液中,大约滴加3min。滴毕,在20℃反应2h后停止反应。用水洗两次,合并有机层,饱和食盐水洗涤,经无水硫酸钠干燥,蒸出二氯甲烷后经柱层析分离得产物,收率为97.5%。1H NMR(400MHz,CDCl3)δ=7.33-7.35(dd,1H,Ar),7.02-7.11(m,2H,Ar),3.88(s,3H,OCH3),3.86(s,3H,OCH3),2,.35(s,4H,CH2)ppm。
实施例9:
将0.5000g 2,3-二甲氧基苯甲酸、0.7750gl-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)和8mL干燥的二氯甲烷依次加入干燥的三颈瓶中。室温下将0.1125g氮杂环丙烷溶于7.5mL二氯甲烷所配成的混合溶液滴加到反应液中,大约滴加3min。滴毕,反应2h后停止反应。用水洗两次,合并有机层,饱和食盐水洗涤,经无水硫酸钠干燥,蒸出二氯甲烷后经柱层析分离得产物,收率为92.6%。1H NMR(400MHz,CDCl3)δ=7.33-7.35(dd,1H,Ar),7.02-7.11(m,2H,Ar),3.88(s,3H,OCH3),3.86(s,3H,OCH3),2,.35(s,4H,CH2)ppm。
步骤四:N-(2-(苯并[d][1,3]二氧杂环戊烯-5-基)乙基)-2,3-二甲基苯甲酰胺的制备
实施例10:
将0.04210g CuBr.SMe2、4mL THF室温下加入干燥的三颈瓶中,N2保护下搅拌。待反应液降至-40℃,滴加步骤2制好的格氏试剂,大约滴加2min,搅拌反应20min。后将0.5818g 1-氮丙啶-2,3-二甲氧基苯基乙酮与4mL四氢呋喃的混合溶液滴加入反应液中,大约滴加2min,滴加完毕后继续反应20min后,停止反应。加入饱和氯化铵淬灭反应,然后用二氯甲烷萃取,合并有机层,饱和食盐水洗涤,经无水硫酸钠干燥,蒸出二氯甲烷后经柱层析分离得产物,收率92.0%。1H NMR(400MHz,CDCl3)δ=8.01(br,s,1H,NH),7.69(dd,1H,HAr),7.13(t,1H,HAr),7.01(d,1H,HAr),6.76-6.70(m,3H,HAr),5.92(s,2H,CH2),3.87(s,3H,OCH3),3.73-3.64(m,5H,OCH3,CH2),2.86(t,2H,CH2)ppm。
实施例11:
将0.04210g CuBr.SMe2、4mL THF室温下加入干燥的三颈瓶中,N2保护下搅拌。待反应液降至-20℃,滴加步骤2制好的格氏试剂,大约滴加2min,搅拌反应20min。后将0.5818g 1-氮丙啶-2,3-二甲氧基苯基乙酮与4mL四氢呋喃的混合溶液滴加入反应液中,大约滴加2min,滴加完毕后继续反应20min后,停止反应。加入饱和氯化铵淬灭反应,然后用二氯甲烷萃取,合并有机层,饱和食盐水洗涤,经无水硫酸钠干燥,蒸出二氯甲烷后经柱层析分离得产物,收率93.2%。1H NMR(400MHz,CDCl3)δ=8.01(br,s,1H,NH),7.69(dd,1H,HAr),7.13(t,1H,HAr),7.01(d,1H,HAr),6.76-6.70(m,3H,HAr),5.92(s,2H,CH2),3.87(s,3H,OCH3),3.73-3.64(m,5H,OCH3,CH2),2.86(t,2H,CH2)ppm。
实施例12:
将0.04210g CuBr.SMe2、4mL THF室温下加入干燥的三颈瓶中,N2保护下搅拌。待反应液降至0℃,滴加步骤2制好的格氏试剂,大约滴加2min,搅拌反应20min。后将0.5818g 1-氮丙啶-2,3-二甲氧基苯基乙酮与4mL四氢呋喃的混合溶液滴加入反应液中,大约滴加2min,滴加完毕后继续反应20min后,停止反应。加入饱和氯化铵淬灭反应,然后用二氯甲烷萃取,合并有机层,饱和食盐水洗涤,经无水硫酸钠干燥,蒸出二氯甲烷后经柱层析分离得产物,收率96.9%。1H NMR(400MHz,CDCl3)δ=8.01(br,s,1H,NH),7.69(dd,1H,HAr),7.13(t,1H,HAr),7.01(d,1H,HAr),6.76-6.70(m,3H,HAr),5.92(s,2H,CH2),3.87(s,3H,OCH3),3.73-3.64(m,5H,OCH3,CH2),2.86(t,2H,CH2)ppm。
以上所述,仅为本发明较佳的具体实施方式,本发明的保护范围不限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,可显而易见地得到的技术方案的简单变化或等效替换均落入本发明的保护范围内。
一种黄连素重要中间体的合成专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。





动态评分
0.0