




专利摘要
本发明公开了一种基于2‑甲基‑5,7‑二溴‑8‑羟基喹啉为配体的单核镝配合物及其制备方法和应用。该配合物的化学式为:[Dy(L)3(H2O)],其中L为2‑甲基‑5,7‑二溴‑8‑羟基喹啉脱去羟基氢原子,带一个单位负电荷;该配合物属于单斜晶系,C2/c空间群。本发明所述配合物的制备方法为:取Dy(NO3)3·6H2O和2‑甲基‑5,7‑二溴‑8‑羟基喹啉,用混合溶剂溶解,调节所得溶液的pH=6.5~7.8,所得混合液于加热条件下反应,即得;其中,所述的混合溶剂为氯仿或氘代氯仿与水的组合物。本发明提供的配合物制备方法简单、成本低廉、重复性好,在低温下表现为慢弛豫磁行为,可用于制备磁性材料。
权利要求
1.基于2-甲基-5,7-二溴-8-羟基喹啉为配体的单核镝配合物,其特征在于:
该配合物的化学式为:[Dy(L)3(H2O)],其中L为2-甲基-5,7-二溴-8-羟基喹啉脱去羟基氢原子,带一个单位负电荷;
该配合物属于单斜晶系,C2/c空间群,晶胞参数为: α=90.00°,β=119.288(5)°,γ=90.00°。
2.权利要求1所述配合物的制备方法,其特征在于:取Dy(NO3)3·6H2O和2-甲基-5,7-二溴-8-羟基喹啉,用混合溶剂溶解,调节所得溶液的pH=6.5~7.8,所得混合液于加热条件下反应,即得;其中,所述的混合溶剂为氯仿或氘代氯仿与水的组合物。
3.根据权利要求2所述的制备方法,其特征在于:所述的混合溶剂中,氯仿或氘代氯仿与水的体积比为1~3:1~3。
4.根据权利要求2所述的制备方法,其特征在于:反应在60~100℃条件下进行。
5.根据权利要求2所述的制备方法,其特征在于:反应的时间为30~80h或80h以上。
6.根据权利要求2所述的制备方法,其特征在于:用三乙胺调节溶液的pH值。
7.权利要求1所述的基于2-甲基-5,7-二溴-8-羟基喹啉为配体的单核镝配合物在制备磁性材料中的应用。
说明书
技术领域
本发明涉及一种基于2-甲基-5,7-二溴-8-羟基喹啉为配体的单核镝配合物及其制备方法和应用,属于磁性材料技术领域。
背景技术
单分子磁体由于结构的特殊性,表现出慢弛豫、量子隧穿以及量子干涉效应等独特的磁现象,使其在量子计算机、超高密度信息存储材料等方面具有潜在的应用前景。
伴随着科研工作者对单分子磁体弛豫机理的深入研究,同时随着表征技术的快速发展,越来越多的具有实用价值的稀土单分子磁体被合成出来,但目前尚未见有化学式为[Dy(C10H6NOBr2)3(H2O)]的磁性材料的相关报道。
发明内容
本发明要解决的技术问题是提供一种新的基于2-甲基-5,7-二溴-8-羟基喹啉为配体的单核镝配合物及其制备方法和应用。
本发明所述的基于2-甲基-5,7-二溴-8-羟基喹啉为配体的单核镝配合物,该配合物的化学式为:[Dy(L)3(H2O)],其中L为2-甲基-5,7-二溴-8-羟基喹啉脱去羟基氢原子,带一个单位负电荷;
该配合物属于单斜晶系,C2/c空间群,晶胞参数为: α=90.00°,β=119.288(5)°,γ=90.00°。
上述技术方案中涉及的2-甲基-5,7-二溴-8-羟基喹啉,可以参考现有文献(如JiantongCui,Synthesis of chlorquinaldol,Zhongguo Yiyao Gongye Zazhi,39(2008),733;或者是Oleg V.Larionov,Direct,catalytic,and regioselective synthesis of 2-alkyl-,aryl-,and alkenyl-substituted N-heterocycles from N-oxides,Organic Letters,16(2014),864-867)进行。
本发明所述的基于2-甲基-5,7-二溴-8-羟基喹啉为配体的单核镝配合物的分子式为C30H20Br6DyN3O4,分子量为1127.41。
本发明所述的基于2-甲基-5,7-二溴-8-羟基喹啉为配体的单核镝配合物的晶体结构数据如下述表1所示,部分键长键角数据如下述表2所示。
表1:[Dy(L)3(H2O)]的晶体学参数
表2:[Dy(L)3(H2O)]的部分键长 键角(°)表
申请人研究发现,本发明所述的基于2-甲基-5,7-二溴-8-羟基喹啉为配体的单核镝配合物的磁学性质表现为慢弛豫磁行为。因此,本发明还包括上述基于2-甲基-5,7-二溴-8-羟基喹啉为配体的单核镝配合物在制备磁性材料中的应用。
上述基于2-甲基-5,7-二溴-8-羟基喹啉为配体的单核镝配合物的制备方法为:取Dy(NO3)3·6H2O和2-甲基-5,7-二溴-8-羟基喹啉,用混合溶剂溶解,调节所得溶液的pH=6.5~7.8,所得混合液于加热条件下反应,即得;其中,所述的混合溶剂为氯仿或氘代氯仿与水的组合物。
上述制备方法中,所述Dy(NO3)3·6H2O和2-甲基-5,7-二溴-8-羟基喹啉的摩尔比为化学计量比,通常为2:1。
上述制备方法中,所述的混合溶剂中,氯仿和水(或者是氘代氯仿和水)的体积比可以为任意配比,优选为1~3:1~3,较佳的选择为1:1、1:2或2:1。所述混合溶剂的用量可根据需要确定,通常以能溶解参加反应的原料为宜,具体地,以1mmol的2-甲基-5,7-二溴-8-羟基喹啉为基准计算,全部原料所用混合溶剂的总用量一般为8~10mL。在具体溶解的步骤中,可将Dy(NO3)3·6H2O与2-甲基-5,7-二溴-8-羟基喹啉分别用混合溶剂溶解,再混合在一起反应,也可将Dy(NO3)3·6H2O与2-甲基-5,7-二溴-8-羟基喹啉混合后再加混合溶剂溶解。申请人在试验过程中发现,仅当混合溶剂为氯仿和水(或者是氘代氯仿和水)的组合时才会有目标配合物生成。
上述制备方法中,可以采用现有常用的碱性物质来调节溶液的pH值,优选是采用三乙胺调节溶液的pH值。本发明所述技术方案中,优选调节溶液的pH=6.8~7.3。
上述制备方法中,通常是将调节pH值后所得的混合液置于容器中,经液氮冷冻后抽至真空,熔封,之后再置于加热条件下反应。所述反应优选是在60~100℃条件下进行,在上述温度条件下进行反应的时间通常为30~80h,也可以是80h以上;优选是将反应时间控制在60~80h。反应更优选是在80~100℃条件下进行。通常采用一端封闭的厚壁硬质玻璃管来盛装调节pH值后所得的混合液。
与现有技术相比,本发明提供了一种新的基于2-甲基-5,7-二溴-8-羟基喹啉为配体的单核镝配合物[Dy(L)3(H2O)]及其制备方法,申请人在研究还发现该配合物在低温下表现为慢弛豫磁行为,可以用于制备磁性材料;此外,该配合物的制备方法简单、成本低廉、重复性好。
附图说明
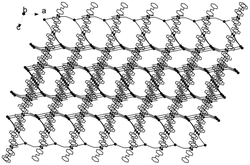
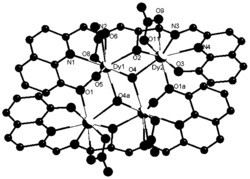
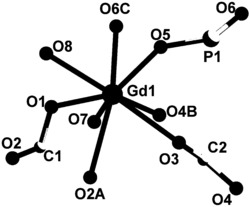
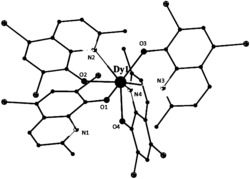
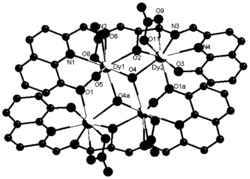
图1为本发明实施例1制得的[Dy(L)3(H2O)]的结构图;
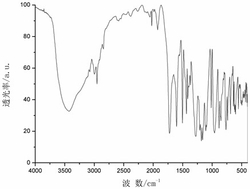
图2为本发明实施例1制得的[Dy(L)3(H2O)]的红外光谱图;
图3为本发明实施例1制得的[Dy(L)3(H2O)]的χm,χmT-T曲线图;
图4为本发明实施例1制得的[Dy(L)3(H2O)]的M-H曲线图;
图5为本发明实施例1制得的[Dy(L)3(H2O)]的交流曲线图。
具体实施方式
下面结合具体实施例对本发明作进一步的详述,以更好地理解本发明的内容,但本发明并不限于以下实施例。
以下各实施例中涉及的2-甲基-5,7-二溴-8-羟基喹啉采用以下方法制备:
将0.1mol 2-甲基-8羟基喹啉加入到100mL冷的冰醋酸中,然后把0.05mol二溴海因分成两份,分批加入到底物的冰醋酸溶液中。每份加完后,继续在冰水浴中反应。3h后反应结束,将反应液倒入到冰水中,不断地搅拌,得到黄色的沉淀。抽滤,滤饼用冰水冲洗三次,常温下干燥,得到黄色的固体,粗品用甲醇重结晶。元素分析(%)(C10H7NOBr2),实验值:C,37.93,H,2.20,N,4.46;理论值:C,37.97,H,2.22,N,4.43。
实施例1
将45mg(约0.2mmol)配体2-甲基-5,7-二溴-8-羟基喹啉和46mg(约0.1mmol)Dy(NO3)3·6H2O加到一端封闭长约18cm的Pyrex管中,加入2mL由CHCl3和H2O组成的混合溶剂(CHCl3和H2O的体积比为1:1),滴加2滴三乙胺(此时溶液pH值为7.2),将Pyrex管抽真空,将其另一端封口。将封好的Pyrex管置于80℃条件下保温反应72h,取出,自然冷却至室温,可观察到Pyrex管底部有黄色条状晶体析出。产率62%。
对上述所得产物进行表征:
1)晶体结构分析:
通过单晶衍射测定表面结构完好的黄色条状晶体以确定其晶体结构,所得晶体结构数据如前述表1所示,键长键角数据如前述表2所示,所得黄色条状晶体的化学结构如图1所示,确定所得黄色条状晶体即为目标配合物[Dy(L)3(H2O)],其中L为2-甲基-5,7-二溴-8-羟基喹啉脱去酚羟基氢原子带一个单位的负电荷,该单核镝配合物的分子式为C30H20Br6DyN3O4,分子量为1127.41。配合物的红外谱图如图2所示。
2)磁学性质测定:
取0.035g本实施例制得的配合物[Dy(L)3(H2O)]碾碎后在磁测量系统上进行磁性测试,得到配合物的交流曲线如图5(包括交流的实部和虚部对温度的曲线)所示,配合物的χM,χMT-T曲线图和M-H曲线图分别如图3和图4所示。
由图5可知,在低温部分,交流的实部和虚部都有明显的频率依赖行为,证实本发明所述的基于2-甲基-5,7-二溴-8-羟基喹啉为配体的单核镝配合物[Dy(L)3(H2O)]在低温下表现为慢弛豫磁行为。
对比例1
重复实施例1,不同的是:用H2O代替实施例1中的混合溶剂,H2O的用量为2mL。
取出Pyrex管并自然冷却至室温后在Pyrex管内并未见有固体产物析出,为澄清的溶液,即没有目标产物生成。
对比例2
重复实施例1,不同的是:将混合溶剂更改为由CHCl2和H2O组成,CHCl2和H2O的体积比为1:1,混合溶剂的总用量不变。
取出Pyrex管并自然冷却至室温后在Pyrex管内并未见有固体产物析出,为澄清的溶液,即没有目标产物生成。
对比例3
重复实施例1,不同的是:将混合溶剂更改为由CH3OH和H2O组成,CH3OH和H2O的体积比为1:1,混合溶剂的总用量不变。
取出Pyrex管并自然冷却至室温后在Pyrex管内并未见有固体产物析出,为澄清的溶液,即没有目标产物生成。
实施例2
重复实施例1,不同的是:将混合溶剂中CHCl3和H2O的体积比更改为1:1,混合溶剂的加入量为1mL。
取出Pyrex管并自然冷却至室温后在Pyrex管底部有黄色条状晶体析出。产率65%。
对所得产物进行结构表征,确定为目标配合物[Dy(L)3(H2O)]。对所得产物的磁学性质表征可知所得产物在低温下表现为慢弛豫磁行为。
实施例3
重复实施例1,不同的是:将混合溶剂中CHCl3和H2O的体积比更改为2:1。
取出Pyrex管并自然冷却至室温后在Pyrex管底部有黄色条状晶体析出。产率60%。
对所得产物进行结构表征,确定为目标配合物[Dy(L)3(H2O)]。对所得产物的磁学性质表征可知所得产物在低温下表现为慢弛豫磁行为。
实施例4
重复实施例1,不同的是:将混合溶剂中CHCl3和H2O的体积比更改为1:2,混合溶剂的加入量为1mL。
取出Pyrex管并自然冷却至室温后在Pyrex管底部有黄色条状晶体析出。产率59%。
对所得产物进行结构表征,确定为目标配合物[Dy(L)3(H2O)]。对所得产物的磁学性质表征可知所得产物在低温下表现为慢弛豫磁行为。
实施例5
重复实施例1,不同的是:将反应时间延长至80h。
取出Pyrex管并自然冷却至室温后在Pyrex管底部有黄色条状晶体析出。产率63%。
对所得产物进行结构表征,确定为目标配合物[Dy(L)3(H2O)]。对所得产物的磁学性质表征可知所得产物在低温下表现为慢弛豫磁行为。
实施例6
重复实施例1,不同的是:将反应温度更改为100℃,将反应时间更改为80h。
取出Pyrex管并自然冷却至室温后在Pyrex管底部有黄色条状晶体析出。产率60%。
对所得产物进行结构表征,确定为目标配合物[Dy(L)3(H2O)]。对所得产物的磁学性质表征可知所得产物在低温下表现为慢弛豫磁行为。
实施例7
重复实施例1,不同的是:
1)将混合溶剂更改为由氘代氯仿和水组成,氘代氯仿和水的体积比为1:1;
2)将反应温度更改为60℃,将反应时间更改为30h。
取出Pyrex管并自然冷却至室温后在Pyrex管底部有黄色条状晶体析出。产率61%。
对所得产物进行结构表征,确定为目标配合物[Dy(L)3(H2O)]。对所得产物的磁学性质表征可知所得产物在低温下表现为慢弛豫磁行为。
实施例8
重复实施例1,不同的是:
1)将混合溶剂更改为由氘代氯仿和水组成,氘代氯仿和水的体积比为2:1;
2)将反应温度更改为40℃,将反应时间更改为60h。
取出Pyrex管并自然冷却至室温后在Pyrex管底部有黄色条状晶体析出。产率59%。
对所得产物进行结构表征,确定为目标配合物[Dy(L)3(H2O)]。对所得产物的磁学性质表征可知所得产物在低温下表现为慢弛豫磁行为。
基于2-甲基-5,7-二溴-8-羟基喹啉为配体的单核镝配合物及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。










动态评分
0.0