

IPC分类号 : C07C49/757,C07C45/78,C07D307/28,C07D317/26,A61K31/122,A61K31/357,A61K31/341,A61P35/00,C12P15/00,C12N1/14,C12R1/645






专利摘要
本发明公开了一种蛇孢菌素类二倍半萜化合物及其制备方法与应用。本发明所提供的化合物的结构式如式I所示,其中,R2为-CH=CH-R3、含氧杂环基或烷基取代的含氧杂环基;所述R3为烃基或含氧杂环基或烷基取代的含氧杂环基;C3与C4之间形成碳碳单键或碳碳双键,当C3与C4之间形成碳碳双键时,R1不存在;当C3与C4之间形成碳碳单键时,R1为羟基。上述化合物是分离得到地衣内生真菌Ulocladiumsp.经过液体发酵,对发酵产物进行分离纯化得到的。经实验证实,该类化合物可作为细胞增殖抑制剂或抗肿瘤剂。 CGMCCNo.5507 2011.11.29
权利要求
1.结构式如式I所示化合物或其药学上可接受的盐、酯、溶剂合物:
其中,R2为-CH=CH-R3、含氧杂环基或烷基取代的含氧环基;所述-CH=CH-R3中的R3为烃基、含氧杂环基或烷基取代的含氧杂环基;
C3与C4之间形成碳碳单键或碳碳双键,当C3与C4之间形成碳碳单键时,R1为羟基,当C3与C4之间形成碳碳双键时,R1不存在。
2.根据权利要求1所述的化合物或其药学上可接受的盐、酯、溶剂合物,其特征在于:所述化合物为下述1)-5)中的任意一种:
1)式I中,C3与C4之间形成碳碳双键、R2为4-甲基-3,4-二羟基-戊烯基的化合物,其结构式如式II所示;
2)式I中,C3与C4之间形成碳碳双键、R2为3-甲氧基-4-甲基-4-羟基-戊烯基的化合物,其结构式如式III所示;
3)式I中,C3与C4之间形成碳碳单键且R1为羟基、R2为3-甲氧基-4-甲基-4-羟基-戊烯基的化合物,其结构式如式IV所示;
4)式I中,C3与C4之间形成碳碳双键、R2为2-(2’,2’,4’,4’-四甲基-1’,3’-二氧戊环基)-乙烯基的化合物,其结构式如式V所示;
5)式I中,C3与C4之间形成碳碳双键、R2为2,2-二甲基-2,5-二氢呋喃环基的化合物,其结构式如式VI所示;
3.制备权利要求1中式I所示化合物的方法,包括下述步骤:
1)将Ulocladium sp.进行发酵培养,得到菌丝体和发酵液的发酵培养物;
2)将所述发酵培养物中的菌丝体和发酵液分离,所得菌丝体破碎后用丙酮水溶液浸提,将丙酮水溶液浸提液浓缩至不含丙酮,水层用乙酸乙酯进行萃取,对萃取液浓缩得菌丝体浸膏;再将所述发酵液浓缩后用乙酸乙酯进行萃取,对萃取液浓缩得发酵液浸膏;将所述菌丝体浸膏和发酵液浸膏合并,即为粗提物;
3)将所述粗提物经硅胶柱层析分离,以石油醚-丙酮为溶剂进行梯度洗脱,收集石油醚-丙酮体积比为10∶1梯度的洗脱物;将收集的洗脱物经凝胶柱层析,以氯仿-甲醇体积比为1∶1的溶剂为洗脱剂;洗脱产物再经反相硅胶柱层析,以甲醇-水为溶剂进行梯度洗脱,收集甲醇-水体积比为70∶30梯度的洗脱产物;将所述洗脱产物经制备HPLC柱分离纯化,以甲醇-水为溶剂进行梯度洗脱,得到式I所示的化合物;
所述经制备HPLC柱分离纯化的过程中,以甲醇-水为溶剂进行梯度洗脱时,起始洗脱溶剂为甲醇-水体积比为20∶80的混合溶剂,终止洗脱溶剂为100%甲醇,洗脱时间为60分钟。
4.根据权利要求3所述的方法,其特征在于:步骤1)中所述发酵培养的培养基组成为:每升培养基含土豆200g、葡萄糖20g,其pH值为6.5;
所述发酵培养的条件为:在25-30℃静置培养14-21天;
所述Ulocladium sp.,它的保藏编号为CGMCC No.5507;
步骤2)中所述丙酮水溶液中丙酮的体积分数60-90%,具体为70%。
5.权利要求1或2所述的化合物或其药学上可接受的盐、酯、溶剂合物在制备真核生物肿瘤细胞增殖抑制剂中的应用或在制备预防和/或治疗肿瘤药物中的应用。
6.根据权利要求5所述的应用,其特征在于:所述真核生物为哺乳动物;所述肿瘤细胞为癌细胞,所述癌细胞具体为宫颈癌细胞、肺癌细胞、口腔表皮样癌细胞或肝癌细胞。
7.权利要求5所述的应用,其特征在于:所述肿瘤为癌;所述癌具体为宫颈癌、肺癌、口腔癌或肝癌。
8.一种药物,它的有效成分为权利要求1或2中所述化合物或其药学上可接受的盐、酯、溶剂合物;所述药物为抑制真核生物肿瘤细胞增殖的药物或预防和/或治疗肿瘤的药物。
9.根据权利要求8所述的药物,其特征在于:所述真核生物为哺乳动物;所述肿瘤细胞为癌细胞;所述癌细胞具体为宫颈癌细胞、肺癌细胞、口腔表皮样癌细胞或肝癌细胞;
所述肿瘤为癌;所述癌具体为宫颈癌、肺癌、口腔癌或肝癌。
10.Ulocladium sp.,其保藏号为CGMCC No.5507。
说明书
技术领域
本发明涉及一种蛇孢菌素类二倍半萜化合物及其制备方法与应用。
背景技术
蛇孢菌素(ophiobolin)类二倍半萜烯化合物已有一些文献报道,该类化合物显示了广谱的抗菌活性,如抗线虫、抗真菌及抗细菌等。据报道,该家族的ophiobolin A能抑制钙离子活化的环核苷酸磷酸二酯酶的活性并导致小鼠白血病细胞L1210细胞的凋亡。
到目前为止已发现该类化合物30余种,该类化合物的结构特征均具有一个5-8-5三环结构。此外,有14、17位形成环氧结构的,如ophiobolin A、ophiobolin J等(Evidente A,Andolfi A,Cimmino A,Vurro M,Fracchiolla M,Charudattan R,Motta A.Ophiobolin E and 8-epi-ophiobolin J produced by Drechslera gigantea,a potential mycoherbicide of weedy grasses.Phytochemistry.2006.67(20):2281-7.);有16、17位双键被氢化而饱和的,如ophiobolins B-D、ophiobolin F、ophiobolin M等(Tsipouras A,Adefarati AA,Tkacz JS,etal.Ophiobolin M and analogues,noncompetitive inhibitors of ivermectin binding with nematocidal activity.Bioorg Med Chem.1996.4(4):531-6.);有5、21位之间形成内酯环或半缩酮的,如ophiobolin A lactone、ophiobolin H、ophiobolin L等(Wei,H;Itoh,T;Kinoshita,M;Nakai,Y;Kurotaki,M;Kobayashi,M;Cytotoxic sesterterpenes,6-epi-ophiobolin G and 6-epi-ophiobolin N,from marine derived fungus Emericella variecolor GF10.Tetrahedron.2004.60(28):6015-9)。
发明内容
本发明的目的是提供一种具有抗肿瘤活性的新化合物及其制备方法。
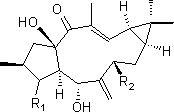
本发明所提供的化合物是首次从地衣内生真菌Ulocladium sp.的发酵物中分离的结构新颖的蛇孢菌素(ophiobolin)类二倍半萜烯化合物,其结构式如式I所示:
其中,R2为-CH=CH-R3、含氧杂环基或烷基取代的含氧环基;所述-CH=CH-R3中的R3为烃基、含氧杂环基或烷基取代的含氧杂环基;
C3与C4之间形成碳碳单键或碳碳双键,当C3与C4之间形成碳碳单键时,R1为羟基,当C3与C4之间形成碳碳双键时,R1不存在。
本发明所述的蛇孢菌素(ophiobolin)类二倍半萜烯化合物具体可为下述1)-5)中的任意一种:
1)式I中,C3与C4之间形成碳碳双键、R2为4-甲基-3,4-二羟基-戊烯基的化合物,其结构式如式II所示(化合物1);
2)式I中,C3与C4之间形成碳碳双键、R2为3-甲氧基-4-甲基-4-羟基-戊烯基的化合物,其结构式如式III所示(化合物2);
3)式I中,C3与C4之间形成碳碳单键且R1为羟基、R2为3-甲氧基-4-甲基-4-羟基-戊烯基的化合物,其结构式如式IV所示(化合物3);
4)式I中,C3与C4之间形成碳碳双键、R2为2-(2’,2’,4’,4’-四甲基-1’,3’-二氧戊环基)-乙烯基的化合物,其结构式如式V所示(化合物4);
5)式I中,C3与C4之间形成碳碳双键、R2为2,2-二甲基-2,5-二氢呋喃环基的化合物,其结构式如式VI所示(化合物5);
上述式I药学上可接受的盐、酯及溶剂合物也属于本发明的保护范围。
本发明所提供的蛇孢菌素(ophiobolin)类二倍半萜烯化合物是从Ulocladium sp.发酵物中提取分离得到的。
具体制备方法如下:
1)将Ulocladium sp.进行发酵培养,得到菌丝体和发酵液的发酵培养物;
2)将所述发酵培养物中的菌丝体和发酵液分离,所得菌丝体破碎后用丙酮水溶液浸提,将丙酮浸提液浓缩至不含丙酮,水层用乙酸乙酯进行萃取,对萃取液浓缩得菌丝体浸膏;再将所述发酵液浓缩后用乙酸乙酯进行萃取,对萃取液浓缩得发酵液浸膏;将所述菌丝体浸膏和发酵液浸膏合并,即为粗提物;
3)将所述粗提物经硅胶柱层析分离,以石油醚-丙酮为溶剂进行梯度洗脱,收集石油醚-丙酮体积比为10∶1梯度的洗脱物;将收集的洗脱物经凝胶柱层析,以氯仿-甲醇体积比为1∶1的溶剂为洗脱剂;洗脱产物再经反相硅胶柱层析,以甲醇-水为溶剂进行梯度洗脱,收集甲醇-水体积比为70∶30梯度的洗脱产物;将所述洗脱产物经制备HPLC分离纯化,以甲醇-水为溶剂进行梯度洗脱,得到式I所示的化合物;
所述经制备HPLC柱分离纯化的过程中,以甲醇-水为溶剂进行梯度洗脱时,起始洗脱溶剂为甲醇-水体积比为20∶80的混合溶剂,终止洗脱溶剂为100%甲醇,洗脱时间为60分钟。
上述步骤1)中所述发酵培养的培养基组成为:每升培养基含土豆200g、葡萄糖20g,其pH值为6.5。所述发酵培养的条件为:在25-30℃静置培养14-21天。
所用的菌株Ulocladium sp.是从云南省紫溪山地衣Everniastrum sp.中分离得到,并于2011年11月29日保藏于中国微生物菌种保藏管理委员会普通微生物中心(简称CGMCC,地址:北京市朝阳区北辰西路1号院3号,中国科学院微生物研究所,邮编100101),保藏号为CGMCC No.5507。
步骤2)所述丙酮水溶液中丙酮的体积分数60-90%,具体可为70%。
本发明所提供的式I化合物除了通过上述微生物发酵培养,再从发酵物中分离纯化得到外;也可由本领域技术人员熟知的化学修饰方法合成获得。
需要特别说明的是,经微生物发酵制备式I化合物的方法中可采用任何能生产该类化合物的微生物,只要能生产该类化合物的微生物均可作为生产菌制备式I化合物。
本发明的另一个目的是提供式I化合物的应用。
本发明所提供的式I化合物的一个应用为其在制备真核生物肿瘤细胞增殖抑制剂中的应用。本发明中所述真核生物具体为哺乳动物。
所述肿瘤细胞为癌细胞;所述癌细胞具体可为宫颈癌细胞、肺癌细胞、口腔表皮样癌细胞或肝癌细胞。
本发明提供的式I化合物的另一个应用为其在制备预防和/或治疗肿瘤药物中的应用。
所述肿瘤为癌;所述癌细胞具体可为宫颈癌、肺癌、口腔癌或肝癌。
此外,式I化合物还可作为抑制肿瘤细胞增殖的低分子生物探针用于生命科学研究。作为探针应用时,式I可溶于甲醇中,也可溶于二甲基亚砜中加以应用。
本发明还保护一种真核生物肿瘤细胞增殖抑制剂,它的有效成分为式I所示的蛇孢菌素(ophiobolin)类二倍半萜烯化合物。
此外,以式I所示的蛇孢菌素(ophiobolin)类二倍半萜烯化合物为有效成分制备的预防和/或治疗肿瘤的药物,也属于本发明的保护范围。
所述预防和/或治疗肿瘤药物可通过注射、喷射、滴鼻、滴眼、渗透、吸收、物理或化学介导的方法导入机体如肌肉、皮内、皮下、静脉、粘膜组织;或是被其他物质混合或包裹后导入机体。
以式I所示的蛇孢菌素(ophiobolin)类二倍半萜烯化合物为活性成分制备的预防和/或治疗的肿瘤药物,需要的时候,在上述药物中还可以加入一种或多种药学上可接受的载体。所述载体包括药学领域常规的稀释剂、赋形剂、填充剂、粘合剂、湿润剂、崩解剂、吸收促进剂、表面活性剂、吸附载体、润滑剂等。
上述预防和/或治疗肿瘤的药物可以制成注射液、片剂、粉剂、颗粒剂、胶囊、口服液、膏剂、霜剂等多种形式。上述各种剂型的药物均可以按照药学领域的常规方法制备。
本发明的发明人通过研究发现地衣内生真菌Ulocladium sp.液体发酵产物经超声破碎后的粗提物有很好的细胞坏死活性,遂对其活性成分进行了研究,采用MTT法测试了化合物对Hela、A549、KB和HepG2的抗肿瘤活性,实验证实本发明的化合物对上述四种肿瘤细胞均有增殖抑制作用。
具体实施方式
下面通过具体实施例对本发明进行说明,但本发明并不局限于此。
下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和生物材料,如无特殊说明,均可从商业途径获得。
实施例1、菌株Ulocladium sp.的分离和鉴定
一、菌株Ulocladium sp.的分离
2005年9月从云南省紫溪山地衣Everniastrum sp.中分离得到的一株菌,命名为菌株651181(L27)。
二、菌株Ulocladium sp.的鉴定
1、形态和生理生化鉴定
菌株在PDA培养基上,28℃培养,菌落生长速度较快,呈褐色。菌丝直径2.5-3um,浅褐色,光滑,有多个分隔。分生孢子单生,褐色至深褐色,成熟时呈倒卵形,90-140*3.5*5.5um,表面光滑,1-3个横膈膜,0-3个纵膈膜。
2、分子鉴定
菌株651181的16SrRNA的序列见序列表的序列1。其与NCBI中登录号为AF229485的Ulocladium alternariae菌株同源性最高,达到92%。根据以上鉴定结果,菌株651181属于Ulocladium sp.。
三、菌株Ulocladium sp.的保藏
细基格孢(Ulocladium sp.)已于2011年11月29日保藏于中国微生物菌种保藏管理委员会普通微生物中心(简称CGMCC,地址:北京市朝阳区北辰西路1号院3号,中国科学院微生物研究所,邮编100101),保藏号为CGMCC No.5507。
实施例2、化合物1-5的发酵生产及分离精制
1、发酵生产
生产菌的发酵培养:按培养微生物的常规方法,取Ulocladium sp.菌种(保藏编号:CGMCC No.5507)适量,接种到PDA斜面培养基上,在28℃培养箱中培养4天。
取斜面培养4天的Ulocladium sp.适量,接种到装有120mL培养液[培养液组成(克/升):4g葡萄糖,10g麦芽糖提取物,4g酵母粉,pH 6.5]的500mL锥型瓶中,在28℃、180rpm条件下摇床培养48小时,获得Ulocladium sp.的种子培养液。
将该种子培养液按5%接种量分别接种于装有200毫升生产培养液[培养基组成(克/升):200g土豆,20g葡萄糖,pH 6.5]的500mL三角烧瓶中,进行为期14天28℃的静置培养发酵,获得菌丝体和发酵液。
2、浸膏的获得
用绢布将菌丝体和发酵液分离。将菌丝体用体积分数为70%的丙酮水溶液破碎浸提三次,将丙酮水溶液浸提液减压浓缩至不含丙酮,所得水层用等体积乙酸乙酷萃取三次,合并乙酸乙酯萃取液减压浓缩,得粗浸膏。发酵液减压浓缩为四分之一体积后,用乙酸乙酯萃取三次,合并菌丝体和发酵液的浸膏,共7.6克。
3、化合物的分离精制
浸膏(7.6g)经硅胶柱层析(200-300目,青岛海洋化工厂)分离,以石油醚-丙酮为溶剂进行梯度洗脱,洗脱比例为100∶1、50∶1、30∶1、20∶1、10∶1、9∶1、8∶2、7∶3和6∶4,每个梯度洗脱溶剂1500毫升,每300毫升收集为一个馏分,通过TLC分析共分成18个馏分,收集石油醚-丙酮体积比为10∶1梯度的洗脱物1.2克;再经凝胶柱层析(Sephadex LH-20,Amersham Biosciences)以氯仿-甲醇1∶1(v/v)为洗脱剂,收集洗脱产物0.8克;然后经反相硅胶柱(YMC)层析,以甲醇-水为溶剂进行梯度洗脱,洗脱比例为30∶70、50∶50、70∶30、80∶20、90∶10和100∶0,每个梯度洗脱溶剂300毫升,每100毫升收集为一个馏分,通过TLC分析共分成8个馏分,收集甲醇-水体积比为70∶30的洗脱产物经制备HPLC分离(RP-18,YMC-Pack,250×10mm Column,5μm)纯化,以甲醇-水梯度洗脱(60分钟内甲醇浓度由20%增加到100%),得到化合物1(6.7mg,tR=38.58min),2(1.7mg,tR=16.36min),3(2.5mg,tR=32.56min),4(8.5mg,tR=27.44min),5(2.7mg,tR=42.14min)。
上述化合物1的结构式如式II所示,化合物2的结构式如式III所示,化合物3的结构式如式IV所示,化合物4的结构式如式V所示,化合物5的结构式如式VI所示。
上述化合物的物理化学常数见表1,结构确证数据见表2-3。
表1、化合物1-5的物理化学常数
表2、化合物1-5的1H NMR数据(500MHz in CDCl3)
a,为600MHz测试
3、化合物1-5的13C NMR数据(125MHz in CDCl3)
a,为600MHz测试;b,信号可以互换
实施例3、抗肿瘤活性的测试
1、实验样品及实验方法
被测样品溶液的配制:测试样品为上述实施例1中分离精制的纯品化合物1-5。准确称取适量样品,用DMSO配制成所需浓度的溶液,供活性测试。
细胞系及细胞的继代培养:活性测试采用人宫颈癌细胞Hela(ATCC CCL-2)、人肺癌细胞A549(ATCC CCL-1885)、口腔表皮样癌细胞KB(ATCC CCL17)和人肝癌细胞HepG2(ATCC HB-8065)细胞系。上述细胞均购自中科院微生物研究所生物资源中心。各种细胞均用含10%胎牛血清的RPMI-1640培养基,在37℃通入5%二氧化碳的培养箱中继代培养。
细胞增殖抑制活性测试方法(MTT法)
本发明采用MTT法,测试评价了被测试样品对癌细胞Hela、A549、KB和HepG2增殖的抑制活性。活细胞线粒体中脱氢酶能够代谢还原黄色的溴化3-(4,5-二甲基噻唑)-2,5-二苯基四氮唑为蓝紫色的不溶于水的甲月替(formazan),formazan的多少可通过酶标仪测定其吸收度求得。由于formazan的量与活细胞数成正比,所以可根据吸收度计算得出活细胞的数目,从而获得药物抑制或杀伤肿瘤细胞的能力。
活性测试时,取对数生长期的肿瘤细胞,用新鲜的RPMI-1640培养基配制成密度为每毫升5×104个细胞的细胞悬液,按每孔200微升接种于96孔板中,在37℃下培养24小时后,每孔加入2微升不同浓度的样品溶液,继续培养72小时。然后加入20μL含MTT的IPMI-1640溶液(5mg/L),再培养4小时,移出150μL培养液后加入150μL DMSO溶解formazan,在540nm处测定其吸收度。
按照IR%=(OD空白对照-OD样品)/OD空白对照×100%式计算每个浓度下的细胞增殖抑制率(IR%),然后利用IC50计算软件计算半数抑制浓度。
2、实验结果
表4、化合物1-5的细胞增殖抑制活性
3结论
化合物1-5对人癌细胞具有抑制作用。因此,本发明制备的式I可作为抗肿瘤剂(即抗肿瘤药物)用于肿瘤的治疗,也可作为细胞增殖抑制的低分子生物探针用于探索生命现象本质的生命科学实验研究中。
蛇孢菌素类二倍半萜化合物及其制备方法与应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。














动态评分
0.0