









专利摘要
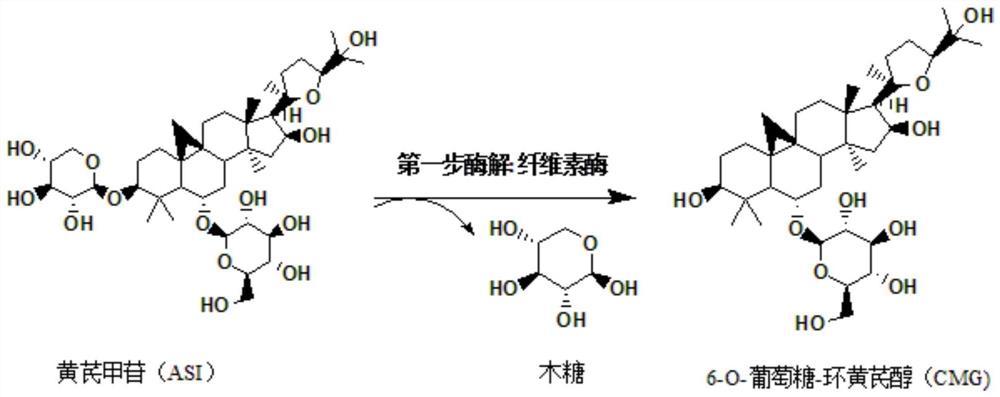
本发明公开了一种环黄芪醇的制备方法及其用途。环黄芪醇的制备方法是以黄芪甲苷或黄芪皂苷为原料,通过氧化、还原、水解、萃取、纯化制得高纯度的环黄芪醇。该方法操作简便、条件温和、产品质量好、收率高。环黄芪醇的用途是,在制备抗癌辅助治疗药物中的应用。以其为药物活性成分制得的抗癌辅助治疗药物,能增强抗癌药物疗效、降低抗癌药物毒性、预防和治疗抗癌药物治疗引起的白细胞降低的抗癌辅助治疗作用。
权利要求
1.一种环黄芪醇的制备方法,包括以下步骤:
A、氧化
将黄芪甲苷加入溶剂配制成黄芪甲苷含量为0.001-0.05mol/L的悬浊液;加酸调节悬浊液的pH至3-6,向悬浊液中加入氧化剂,在5-35℃下避光反应12-96小时,然后加入乙二醇终止氧化,其中黄芪甲苷与氧化剂的摩尔比为1:2-1:15;随后加碱调pH至7-10,减压浓缩至无醇味,再经过滤或萃取得到氧化产物;
B、还原
将A步得到的氧化产物加入溶剂分散成溶液或悬浊液;加入的溶剂与A步的黄芪甲苷的质量比为1:0.7-1:40;然后加入硼氢化钠或硼氢化钾作为还原剂,还原反应12-48h后,再加入乙酸或乙酸水溶液调节反应液的pH至4-6酸性以终止还原反应;其中还原剂与A步的黄芪甲苷的摩尔比为1:2-1:8;
C、水解
向步骤B的反应液加入强酸,在10-60℃下静置或搅拌6-96h;加入的强酸与反应液的质量比为0.5-3:100;
D、萃取;
向步骤C得到的反应液中加碱、调pH至中性,再采用与反应液等体积的乙酸乙酯萃取3-5次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用正相硅胶柱层析、反相硅胶柱层析、重结晶中的一种或多种方法进行纯化,即得环黄芪醇。
2.一种环黄芪醇的制备方法,包括以下步骤:
A、氧化
将黄芪皂苷加入溶剂配制成黄芪皂苷含量为0.6-50g/L的悬浊液;加酸调节悬浊液的pH至3-6,再向悬浊液中加入氧化剂,在5-35℃下避光反应12-96小时,然后加入乙二醇终止氧化,其中黄芪皂苷与氧化剂的质量比为3:1-1:5;随后加碱调pH至7-10,减压浓缩至无醇味,再经过滤或萃取得到氧化产物;
B、还原
将A步得到的氧化产物加入溶剂分散成溶液或悬浊液,加入的溶剂与A步的黄芪皂苷的质量比为1:0.6-1:50;然后加入硼氢化钠或硼氢化钾作为还原剂,还原反应12-48h后,再加入乙酸或乙酸水溶液调节反应液的pH至4-6酸性以终止还原反应;其中还原剂与A步的黄芪皂苷的质量比为1:1.5-1:20;
C、水解
向步骤B的反应液加入强酸,在10-60℃下静置或搅拌反应6-96h;加入的强酸与反应液的质量比为0.5-3:100;
D、萃取
向步骤C得到的反应液中加入碱,调pH至中性,采用与反应液等体积的乙酸乙酯萃取3-5次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用正相硅胶柱层析、反相硅胶柱层析、重结晶中的一种或多种方法进行纯化,即得环黄芪醇。
3.根据权利2要求所述的环黄芪醇的制备方法,其特征在于,所述步骤A的黄芪皂苷为环黄芪醇类皂苷的含量不低于50%的黄芪皂苷。
4.根据权利要求1或2所述的环黄芪醇的制备方法,其特征在于,所述步骤A中的溶剂选用水、甲醇、乙醇中的一种或一种以上的混合液。
5.根据权利1或2要求所述的环黄芪醇的制备方法,其特征在于,所述步骤A中的氧化剂为高碘酸、高碘酸钠或高碘酸钾。
6.根据权利要求1或2所述的环黄芪醇的制备方法,其特征在于,所述步骤A中的萃取的具体操作是:加入与减压浓缩后的反应液等体积的正丁醇萃取2-4次;合并正丁醇萃取液,水洗2次,再蒸干正丁醇。
7.根据权利要求1或2所述的环黄芪醇的制备方法,其特征在于,所述步骤B中的溶剂为体积分数是0-50%的甲醇水溶液或乙醇水溶液。
8.根据权利要求1或2所述的环黄芪醇的制备方法,其特征在于,所述步骤C中加入的强酸为硫酸、盐酸或硝酸。
9.一种权利1或2要求所述的制备方法得到的环黄芪醇的用途是,环黄芪醇在制备抗癌辅助治疗药物中的应用。
说明书
技术领域
本发明涉及一种环黄芪醇的制备方法及其用途,属于医药化工领域
背景技术
癌症是危害人类健康的三大疾病之一,其发病率及死亡率在某些国家已上升到第1位,在我国每年大约有350万人患癌,250万人死于癌症,而且还在不断的增长,并逐渐呈年轻化趋势。癌症每年给我国造成的直接经济损失逾千亿元。因此,研制抗癌药物是改善人类生活质量的一个重要方式,也是是广大医药工作者面临的重大课题之一。目前开发成功的很多抗癌药物,虽然有较明显治疗效果,但其毒副反应也显而易见,它在杀伤癌细胞的同时,也杀害了人体正常细胞,伴随着药物治疗而来的,是患者胃肠道功能紊乱、肝肾功能的损伤、白细胞数目减少和免疫力降低等毒副作用;使抗癌药物的临床应用受到限制。因此,目前抗癌药物研究,一方面继续寻找杀死癌细胞的治疗药物外,开发增强抗癌药物的疗效,降低其毒副作用为主要功效的抗癌辅助药物也越来越受到重视。
黄芪皂苷,是黄芪属(Astragalus)植物中的一类活性成分,是黄芪药效物质基础重要药组成之一,是一类对人类健康有显著保健作用的天然活性成分,具有调节机体免疫、抗衰老、抗肿瘤、抗病毒、保护心脑血管、保护肝肾、促进骨髓造血等作用。当其与抗肿瘤化疗药物配合使用时,能较好的改善人体对化疗药物的耐受性,提高癌症患者的生存质量;但由于黄芪皂苷的相对分子质量较大、具有溶解惰性等原因使其在体内的相对生物利用度不足,限制了其作为药物的开发和推广。
发明内容
本发明的第一目的是提供一种环黄芪醇的制备方法,该种方法反应温和、操作简便、收率高、产品质量好、便于大规模工业化生产。
本发明的第二目的是提供环黄芪醇的用途,即在制备抗癌辅助治疗药物中的应用。以其为药物活性成分制得的抗癌辅助治疗药物,能增强抗癌药物疗效、降低抗癌药物毒性、预防和治疗抗癌药物治疗引起的白细胞降低的抗癌辅助治疗作用。
本发明实现其第一目的,所采用的第一种技术方案是,一种环黄芪醇的制备方法,包括以下步骤:
A、氧化
将黄芪甲苷加入溶剂配制成黄芪甲苷含量为0.001-0.05mol/L的悬浊液;加酸调节悬浊液的pH至3-6,向悬浊液中加入氧化剂,在5-35℃下避光反应12-96小时,然后加入乙二醇终止氧化,其中黄芪甲苷与氧化剂的摩尔比为1:2-1:15;随后加碱调pH至7-10,减压浓缩至无醇味,再经过滤或萃取得到氧化产物;
B、还原
将A步得到的氧化产物加入溶剂分散成溶液或悬浊液;加入的溶剂与A步的黄芪甲苷的质量比为1:0.7-1:40;然后加入硼氢化钠或硼氢化钾作为还原剂,还原反应12-48h后,再加入乙酸或乙酸水溶液调节反应液的pH至4-6酸性以终止还原反应;其中还原剂与A步的黄芪甲苷的摩尔比为1:2-1:8;
C、水解
向步骤B的反应液加入强酸,在10-60℃下静置或搅拌反应6-96h;加入的强酸与反应液的质量比为0.5-3:100;
D、萃取;
向步骤C得到的反应液中加碱、调pH至中性,再采用与反应液等体积的乙酸乙酯萃取3-5次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用正相硅胶柱层析、反相硅胶柱层析、重结晶中的一种或多种方法进行纯化,即得环黄芪醇。
本发明实现其第一目的,所采用的第二种技术方案是,一种环黄芪醇的制备方法,包括以下步骤:
A、氧化
将黄芪皂苷加入溶剂配制成黄芪皂苷含量为0.6-50g/L的悬浊液;加酸调节悬浊液的pH至3-6,再向悬浊液中加入氧化剂,在5-35℃下避光反应12-96小时,然后加入乙二醇终止氧化,其中黄芪皂苷与氧化剂的质量比为3:1-1:5;随后加碱调pH至7-10,减压浓缩至无醇味,再经过滤或萃取得到氧化产物;
B、还原
将A步得到的氧化产物加入溶剂分散成溶液或悬浊液,加入的溶剂与A步的黄芪皂苷的质量比为1:0.6-1:50;然后加入硼氢化钠或硼氢化钾作为还原剂,还原反应12-48h后,再加入乙酸或乙酸水溶液调节反应液的pH至4-6酸性以终止还原反应;其中还原剂与A步的黄芪皂苷的质量比为1:1.5-1:20;
C、水解
向步骤B的反应液加入强酸,在10-60℃下静置或搅拌反应6-96h;加入的强酸与反应液的质量比为0.5-3:100;
D、萃取
向步骤C得到的反应液中加入碱,调pH至中性,采用与反应液等体积的乙酸乙酯萃取3-5次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用正相硅胶柱层析、反相硅胶柱层析、重结晶中的一种或多种方法进行纯化,即得环黄芪醇。
与现有技术相比,本发明的有益成果是:
一、黄芪甲苷或黄芪皂苷通过液相中的氧化、还原、水解、萃取和纯化反应,使黄芪甲苷或黄芪皂苷降解得到较小分子量的环黄芪醇,并有效去除其它成分,获得的环黄芪醇的品质高。测试表明本发明方法制备的环黄芪醇的纯度在95%以上。
二、整个制备过程均在液相中进行、最高温度不超过80度,其工艺条件温和、收率高、操作简便、对操作人员技术要求低,适合工业化推广。
上述步骤A的黄芪皂苷为环黄芪醇类皂苷的含量不低于50%的黄芪皂苷。
当环黄芪醇类皂苷的含量低于50%时,混杂有较多的黄酮苷等成分,在反应时会发生较多的副反应,从而影响制备物的纯度。
上述步骤A中的溶剂选用水、甲醇、乙醇中的一种或一种以上的混合液。
这三种溶剂能较好的溶解黄芪皂苷,且其价廉易得。
上述步骤A中的氧化剂为高碘酸、高碘酸钠或高碘酸钾。
这三种氧化剂既能对黄芪皂苷中的糖部分进行氧化使其开链,以便于后续的水解去除,又不影响苷元(环黄芪醇)部分,从而能很好地提取出环黄芪醇。
上述步骤A中的萃取的具体操作是:加入与减压浓缩后的反应液等体积的正丁醇萃取2-4次;合并正丁醇萃取液,水洗2次,再蒸干正丁醇。
使用正丁醇萃取,能够有效的去除步骤A中产生的小分子杂质,另外可去除未完全反应的氧化剂。
上述步骤B中的溶剂为体积分数为0-50%的甲醇水溶液或乙醇水溶液。
使用体积分数为0-50%的甲醇水溶液或乙醇水溶液,能够较好的溶解和分散A步的氧化产物,且对D步的萃取无影响。
上述步骤C中加入的强酸为硫酸、盐酸或硝酸。
这三种强酸价廉易得,并能很好的进攻糖苷键使其断裂,从而水解得到环黄芪醇。下面结合具体实施方式对本发明作进一步的详细说明。
本发明实现其第二发明目的所采用的技术方案是,上述制备方法得到的环黄芪醇的用途是,环黄芪醇在制备抗癌辅助治疗药物中的应用。
上述用途的具体应用方法是:将环黄芪醇作为活性成分,添加制药辅剂采用药物制剂技术,制成抗癌辅助治疗用的固体口服制剂或液体口服制剂。
所述的固体口服制剂优选为普通片剂、分散片、肠溶片、颗粒、胶囊、滴丸、散剂或缓控释制剂。所述的缓控释制剂优选为缓控释片剂、颗粒或胶囊。
所述的液体口服制剂优选为口服液或乳剂。
采用本发明方法制得的环黄芪醇,其品质高,纯度在95%以上,将其作为药效成分制成抗癌辅助治疗药物,药效实验表明其抗癌辅助治疗效果显著:
与单独使用抗癌药物相比,在对小鼠H22肝癌和S180肉瘤的抑制率增加20%以上;同时明显改善由于单独使用抗癌药物引起的胸腺指数、脾指数、白细胞数下降,与单独使用抗癌药物相比,这些参数改善50%以上。在相同效果下,作为抗癌辅助药物,其用药量仅为黄芪皂苷的30%及以下。
高纯度环黄芪醇用于抗癌辅助药物效果好的可能机理是:与黄芪皂苷相比,环黄芪醇具有相对较小的分子质量和较强的亲脂性,其在生物膜渗透和胃肠道吸收方面具有优势,有更好的生物利用度,从而有更好的抗癌辅助治疗效果。
具体实施方式
实施例1
A、氧化
取780mg(约1mmol)黄芪甲苷作为原料,往其中加入水配置成黄芪甲苷含量为0.01mol/L的悬浊液,加盐酸调节悬浊液的pH至3-4,向悬浊液中加入氧化剂——高碘酸钠在20℃下避光反应48小时,然后加入乙二醇终止氧化,其中黄芪甲苷与氧化剂的摩尔比为1:10;随后加Na2CO3调pH至7-8,减压浓缩至无醇味,加入等体积正丁醇萃取3次,合并正丁醇萃取部分,水洗2次,取正丁醇部分,蒸去正丁醇,得到氧化产物。
B、还原
将A步得到的氧化产物加入30%的甲醇水溶液分散成悬浊液,加入的30%甲醇水溶液与A步的黄芪甲苷的质量比为1:0.7;然后加入硼氢化钠作为还原剂,还原反应24h后,加入50%乙酸至反应液pH到4-6终止反应;其中加入的还原剂与A步的黄芪甲苷的摩尔比为1:2。
C、水解
向步骤B的反应液加入硫酸,使加入的硫酸与反应液的质量比为0.5:100,在50℃下静置反应48h;
D、萃取;
向步骤C得到的反应液中加入K2CO3,调pH至中性,再采用与反应液等体积乙酸乙酯萃取3次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用重结晶方法纯化:用适量的CH2Cl2-丙酮溶解后,然后置于10℃条件下放置24h,析晶,过滤干燥后即得纯度大于90%的环黄芪醇单体化合物。
经检测本例制得的环黄芪醇,其纯度为99%。
实施例2
A、氧化
取1.5g(约2mmol)黄芪甲苷作为原料,往其中加入水配置成黄芪甲苷含量为0.001mol/L的悬浊液,加硫酸调节悬浊液的pH至4-5,向悬浊液中加入氧化剂——高碘酸在5℃下避光反应96小时,然后加入乙二醇终止氧化,其中黄芪甲苷与氧化剂的摩尔比为1:2;随后加NaOH调pH至8-9,过滤反应液得到滤饼,用水润洗滤饼多次后,得到氧化产物
B、还原
将A步得到的氧化产物用水分散成悬浊液,加入的水与A步的黄芪甲苷的质量比为1:20;然后加入硼氢化钠作为还原剂,还原反应12h后,加入50%乙酸至反应液pH到4-6终止反应;其中加入的还原剂与A步的黄芪甲苷的摩尔比为1:4。
C、水解
向步骤B的反应液加入盐酸,使加入的盐酸与反应液的质量比为2:100,在30℃下搅拌反应72h;
D、萃取;
向步骤C得到的反应液中加入NaHCO3,调pH至中性,再采用与反应液等体积乙酸乙酯萃取4次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用重结晶方法纯化:用适量的石油醚-丙酮溶解后,然后置于4℃条件下放置24h,析晶,过滤干燥后即得纯度大于90%的环黄芪醇单体化合物。
经检测本例制得的环黄芪醇,其纯度为98%。
实施例3
A、氧化
取800mg(1mmol)黄芪甲苷作为原料,往其中加入20%甲醇水溶液配置成黄芪甲苷含量为0.05mol/L的悬浊液,加磷酸调节悬浊液的pH至5-6,向悬浊液中加入氧化剂——高碘酸在25℃下避光反应48小时,然后加入乙二醇终止氧化,其中黄芪甲苷与氧化剂的摩尔比为1:15;随后加KHCO3调pH至7-8,减压浓缩至无醇味,加入等体积正丁醇萃取3次,合并正丁醇萃取部分,水洗2次,取正丁醇部分,蒸去正丁醇,得到氧化产物。
B、还原
将A步得到的氧化产物加入50%的甲醇水溶液分散成溶液,加入的50%甲醇水溶液与A步的黄芪甲苷的质量比为1:40;然后加入硼氢化钠作为还原剂,还原反应24h后,加入50%乙酸至反应液pH到4-6终止反应;其中加入的还原剂与A步的黄芪甲苷的摩尔比为1:8。
C、水解
向步骤B的反应液加入硫酸,使加入的硫酸与反应液的质量比为0.5:100,在60℃下搅拌反应12h;
D、萃取;
向步骤C得到的反应液中加氨水,调pH至中性,再采用与反应液等体积乙酸乙酯萃取5次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用重结晶方法纯化:用适量的丙酮溶解后,然后置于-4℃条件下放置24h,析晶,过滤干燥后即得纯度大于90%的环黄芪醇单体化合物。
经检测本例制得的环黄芪醇,其纯度为98%。
实施例4:
A、氧化
取78.5mg(0.1mmol)黄芪甲苷作为原料,往其中加入50%甲醇水溶液配置成黄芪甲苷含量为0.05mol/L的悬浊液,加甲酸调节悬浊液的pH至3-4,向悬浊液中加入氧化剂——高碘酸钾在20℃条件下避光反应12小时,然后加入乙二醇终止氧化,其中黄芪甲苷与氧化剂的摩尔比为1:5;随后加Na2CO3调pH至7-8,过滤反应液得到滤饼,用水润洗滤饼多次后,得到氧化产物。
B、还原
将A步得到的氧化产物加入20%的乙醇水溶液分散成悬浊液,加入的20%乙醇水溶液与A步的黄芪甲苷的质量比为1:10;然后加入硼氢化钠作为还原剂,还原反应12h后,加入50%乙酸至反应液pH到4-5终止反应;其中加入的还原剂与A步的黄芪甲苷的摩尔比为1:8。
C、水解
向步骤B的反应液加入盐酸,使加入的盐酸与反应液的重量比为1:100,在40℃下搅拌反应12h。
D、萃取;
向步骤C得到的反应液中加入KHCO3,调pH至中性,再采用与反应液等体积乙酸乙酯萃取3次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用重结晶方法纯化:用适量的甲醇溶解后,滴加水使水的比例为10%(v/v),然后置于0℃冰水浴条件下放置24h,析晶,过滤干燥后即得纯度大于90%的环黄芪醇单体化合物。
经检测本例制得的环黄芪醇,其纯度为97%
实施例5:
A、氧化
取1.5g(约2mmol)黄芪甲苷作为原料,往其中加入乙醇配置成黄芪甲苷含量为0.05mol/L的悬浊液,加磷酸调节悬浊液的pH至5-6,向悬浊液中加入氧化剂——高碘酸钾在5℃下避光反应96小时,然后加入乙二醇终止氧化,其中黄芪甲苷与氧化剂的摩尔比为1:13;随后加NaHCO3调pH至8-9,减压浓缩至无醇味,过滤反应液得到滤饼,用水润洗滤饼几次后,得到氧化产物。
B、还原
将A步得到的氧化产物加入水溶解分散成悬浊液,加入的水与A步的黄芪甲苷的质量比为1:10;然后加入硼氢化钠作为还原剂,还原反应24h后,加入乙酸至反应液pH到4-5终止反应;其中加入的还原剂与A步的黄芪甲苷的摩尔比为1:2。
C、水解
向步骤B的反应液加入硝酸,使加入的硝酸与反应液的质量比为1:100,在15℃下静置反应96h;
D、萃取;
向步骤C得到的反应液中加氨水,调pH至中性,再采用与反应液等体积乙酸乙酯萃取3次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用正相硅胶柱层析纯化:洗脱剂选用石油醚:乙酸乙酯=5:1~1:5,通过薄层层析法观察,合并环黄芪醇纯度较高的流份,浓缩除去溶剂,即得纯度大于90%的环黄芪醇。
经检测本例制得的环黄芪醇,其纯度为96%。
实施例6:
A、氧化
取400mg(约0.5mmol)黄芪甲苷作为原料,往其中加入40%乙醇水溶液配置成黄芪甲苷含量为0.005mol/L的悬浊液,加盐酸调节悬浊液的pH至3-4,向悬浊液中加入氧化剂——高碘酸钾在5℃下避光反应96小时,其中黄芪甲苷与氧化剂的摩尔比为1:4;然后加入乙二醇终止氧化;随后加NaOH调pH至9-10,减压浓缩至无醇味,加入等体积正丁醇萃取2次,合并正丁醇萃取部分,水洗2次,取正丁醇部分,蒸去正丁醇,得到氧化产物。
B、还原
将A步得到的氧化产物加入50%乙醇水溶液分散成溶液,加入的乙醇水溶液与A步的黄芪甲苷的质量比为1:15;然后加入硼氢化钠作为还原剂,还原反应24h后,加入乙酸至反应液pH到4-5终止反应;其中加入的还原剂与A步的黄芪甲苷的摩尔比为1:5。
C、水解
向步骤B的反应液加入盐酸,使加入的硫酸与反应液的质量比为3:100,在15℃下搅拌反应96h;
D、萃取;
向步骤C得到的反应液中加KOH,调pH至中性,再采用与反应液等体积乙酸乙酯萃取3次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用正相硅胶柱层析结合重结晶纯化:洗脱剂选用石油醚:丙酮=5:1~1:3,通过薄层层析法观察,合并环黄芪醇纯度较高的流份,浓缩除去溶剂,用适量氯仿-甲醇溶解,置于4℃下静置24h,析晶,过滤干燥后即得纯度大于90%的环黄芪醇。
经检测本例制得的环黄芪醇,其纯度为98%。
实施例7:
A、氧化
取6g(约4mmol)黄芪甲苷作为原料,往其中加入水配置成黄芪甲苷含量为0.001mol/L的悬浊液,加盐酸调节悬浊液的pH至3-4,向悬浊液中加入氧化剂——高碘酸钾在15℃下避光反应72小时,然后加入乙二醇终止氧化,其中黄芪甲苷与氧化剂的摩尔比为1:10;随后加NaHCO3调pH至8-9,减压浓缩至无醇味,加入等体积正丁醇萃取4次,合并正丁醇萃取部分,水洗2次,取正丁醇部分,蒸去正丁醇,得到氧化产物。
B、还原
将A步得到的氧化产物加入10%甲醇水溶液分散成悬浊液,加入的10%甲醇水溶液与A步的黄芪甲苷的质量比为1:30;然后加入硼氢化钾作为还原剂,还原反应48h后,加入乙酸至反应液pH到5-6终止反应;其中加入的还原剂与A步的黄芪甲苷的摩尔比为1:2。
C、水解
向步骤B的反应液加入硫酸,使加入的硫酸与反应液的质量比为3:100,在10℃下搅拌反应6h;
D、萃取;
向步骤C得到的反应液中加KOH,调pH至中性,再采用与反应液等体积乙酸乙酯萃取5次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用正相硅胶柱层析纯化:洗脱剂选用乙酸乙酯:甲醇=100:0.1~10:1,通过薄层层析法观察,合并环黄芪醇纯度较高的流份,浓缩除去溶剂,即得纯度大于90%的环黄芪醇。
经检测本例制得的环黄芪醇,其纯度为95%。
实施例8:
A、氧化
取1.5g(约2mmol)黄芪甲苷作为原料,往其中加入体积比为1:1的甲醇乙醇溶液配置成黄芪甲苷含量为0.01mol/L的悬浊液,加乙酸调节悬浊液的pH至3-4,向悬浊液中加入氧化剂——高碘酸在25℃下避光反应48小时,然后加入乙二醇终止氧化,其中黄芪甲苷与氧化剂的摩尔比为1:7;随后加KOH调pH至9-10,减压浓缩至无醇味,加入等体积正丁醇萃取4次,合并正丁醇萃取部分,水洗2次,取正丁醇部分,蒸去正丁醇,得到氧化产物。
B、还原
将A步得到的氧化产物加入5%乙醇水溶液分散成悬浊液,加入的5%乙醇水溶液与A步的黄芪甲苷的质量比为1:40。然后加入硼氢化钾作为还原剂,还原反应24h后,加入乙酸至反应液pH到4-5终止反应;其中加入的还原剂与A步的黄芪甲苷的摩尔比为1:8。
C、水解
向步骤B的反应液加入硫酸,使加入的硫酸与反应液的质量比为1:100,在60℃下静置反应36h;
D、萃取;
向步骤C得到的反应液中加NaOH,调pH至中性,再采用与反应液等体积乙酸乙酯萃取4次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用反相硅胶柱层析纯化:洗脱剂选用甲醇:水=50:50~100:0,通过薄层层析法观察,合并环黄芪醇纯度较高的流份,浓缩除去溶剂,即得纯度大于90%的环黄芪醇。
经检测本例制得的环黄芪醇,其纯度为96%。
实施例9:
A、氧化
取1.5g(约2mmol)黄芪甲苷作为原料,往其中加入10%乙醇水溶液配置成黄芪甲苷含量为0.03mol/L的悬浊液,加磷酸调节悬浊液的pH至4-5,向悬浊液中加入氧化剂——高碘酸在25℃下避光反应48小时,然后加入乙二醇终止氧化,其中黄芪甲苷与氧化剂的摩尔比为1:3;随后加KOH调pH至9-10,减压浓缩至无醇味,过滤反应液得到滤饼,用水润洗滤饼几次后,得到氧化产物。
B、还原
将A步得到的氧化产物加入40%乙醇水溶液分散成悬浊液,加入的40%乙醇水溶液与A步的黄芪甲苷的质量比为1:1。然后加入硼氢化钾作为还原剂,还原反应48h后,加入乙酸至反应液pH到4-5终止反应;其中加入的还原剂与A步的黄芪甲苷的摩尔比为1:3。
C、水解
向步骤B的反应液加入硝酸,使加入的硝酸与反应液的质量比为2:100,在20℃下搅拌反应60h;
D、萃取;
向步骤C得到的反应液中加氨水,调pH至中性,再采用与反应液等体积乙酸乙酯萃取3次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用反相硅胶柱层析纯化,洗脱剂选用乙腈:水=30:70~80:20,通过薄层层析法观察,合并环黄芪醇纯度较高的流份,浓缩除去溶剂,即得纯度大于90%的环黄芪醇。
经检测本例制得的环黄芪醇,其纯度为96%。
实施例10:
A、氧化
取3g黄芪皂苷作为原料,往其中加入20%乙醇水溶液配置成黄芪皂苷含量为0.6g/L的悬浊液,加乙酸调节悬浊液的pH至3-4,向悬浊液中加入氧化剂——高碘酸在20℃下避光反应24小时,然后加入乙二醇终止氧化,其中黄芪皂苷与氧化剂的质量比为3:1;随后加NaOH调pH至8-9,减压浓缩至无醇味,加入等体积正丁醇萃取3次,合并正丁醇萃取部分,水洗2次,取正丁醇部分,蒸去正丁醇,得到氧化产物。
B、还原
将A步得到的氧化产物加入10%甲醇水溶液分散成悬浊液,加入的10%甲醇水溶液与A步的黄芪皂苷的质量比为1:10;然后加入硼氢化钾作为还原剂,还原反应12h后,加入30%乙酸至反应液pH到5-6终止反应;其中加入的还原剂与A步的黄芪皂苷的质量比为1:1.5。
C、水解
向步骤B的反应液加入硫酸,使加入的硫酸与反应液的质量比为1:100,在60℃下搅拌反应6h。
D、萃取;
向步骤C得到的反应液中加KOH,调pH至中性,再采用与反应液等体积乙酸乙酯萃取3次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用重结晶方法纯化:用适量的乙酸乙酯-乙醇溶解后,然后置于-10℃条件下放置24h,析晶,过滤干燥后即得纯度大于90%的环黄芪醇单体化合物。
经检测本例制得的环黄芪醇,其纯度为96%。
实施例11:
A、氧化
取3g黄芪皂苷作为原料,往其中加入水配置成黄芪皂苷含量为30g/L的悬浊液,加甲酸调节悬浊液的pH至4-5,向悬浊液中加入氧化剂——高碘酸钠在25℃下避光反应12小时,然后加入乙二醇终止氧化,其中黄芪皂苷与氧化剂的质量比为1:1;随后加氨水调pH至8-9,减压浓缩至无醇味,加入等体积正丁醇萃取4次,合并正丁醇萃取部分,水洗2次,取正丁醇部分,蒸去正丁醇,得到氧化产物。
B、还原
将A步得到的氧化产物加入50%甲醇水溶液分散成悬浊液,加入的50%甲醇水溶液与A步的黄芪皂苷的质量比为1:0.6。然后加入硼氢化钠作为还原剂,还原反应24h后,加入乙酸至反应液pH到4-5终止反应;其中加入的还原剂与A步的黄芪皂苷的质量比为1:10。
C、水解
向步骤B的反应液加入硫酸,使加入的硫酸与反应液的质量比为2:100,在50℃下搅拌反应12h。
D、萃取;
向步骤C得到的反应液中加NaOH,调pH至中性,再采用与反应液等体积乙酸乙酯萃取3次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用重结晶方法纯化:用适量的CH2Cl2-甲醇溶解后,然后置于-4℃条件下放置24h,析晶,过滤干燥后即得纯度大于90%的环黄芪醇单体化合物。
经检测本例制得的环黄芪醇,其纯度为95%。
实施例12:
A、氧化
取3g黄芪皂苷作为原料,往其中加入30%乙醇水溶液配置成黄芪皂苷含量为10g/L的悬浊液,加磷酸调节悬浊液的pH至5-6,向悬浊液中加入氧化剂——高碘酸钠在20℃下避光反应24小时,然后加入乙二醇终止氧化,其中黄芪皂苷与氧化剂的质量比为1:5;随后加NaOH调pH至8-9,减压浓缩至无醇味,加入等体积正丁醇萃取3次,合并正丁醇萃取部分,水洗2次,取正丁醇部分,蒸去正丁醇,得到氧化产物。
B、还原
将A步得到的氧化产物加入水分散成悬浊液,加入的水与A步的黄芪皂苷的质量比为1:50;然后加入硼氢化钠作为还原剂,还原反应24h后,加入30%乙酸水溶液至反应液pH到5-6终止反应;其中加入的还原剂与A步的黄芪皂苷的质量比为1:20。
C、水解
向步骤B的反应液加入盐酸,使加入的盐酸与反应液的质量比为3:100,在20℃下静置反应72h。
D、萃取;
向步骤C得到的反应液中加NaOH,调pH至中性,再采用与反应液等体积乙酸乙酯萃取3次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用正相硅胶柱层析结合重结晶纯化:用200~300目的正相硅胶柱层析纯化,样品与硅胶的质量比为1:7,洗脱剂选用CH2Cl2:丙酮=7:1~1:1,通过薄层层析法观察,合并环黄芪醇纯度较高的流份,浓缩除去溶剂,用适量石油醚-丙酮溶解,置于0℃下放置48h,析晶,过滤干燥即得纯度大于90%的环黄芪醇。
经检测本例制得的环黄芪醇,其纯度为97%。
实施例13:
A、氧化
取3g黄芪皂苷作为原料,往其中加入水配置成黄芪皂苷含量为50g/L的悬浊液,加硫酸调节悬浊液的pH至4-5,向悬浊液中加入氧化剂——高碘酸在25℃下避光反应12小时,然后加入乙二醇终止氧化,其中黄芪皂苷与氧化剂的质量比为1:3;随后加氨水调pH至7-8,减压浓缩至无醇味,过滤反应液得到滤饼,用水润洗滤饼几次后,得到氧化产物。
B、还原
将A步得到的氧化产物加入30%乙醇水溶液分散成溶液,加入的30%乙醇水溶液与A步的黄芪皂苷的质量比为1:5。然后加入硼氢化钠作为还原剂,还原反应24h后,加入乙酸至反应液pH到4-5终止反应;其中加入的还原剂与A步的黄芪皂苷的质量比为1:6。
C、水解
向步骤B的反应液加入硝酸,使加入的硝酸与反应液的质量比为1.5:100,在60℃下静置反应36h。
D、萃取;
向步骤C得到的反应液中加NaHCO3,调pH至中性,再采用与反应液等体积乙酸乙酯萃取3次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用正相硅胶柱层析纯化:洗脱剂选用CH2Cl2:甲醇=100:1~10:1,通过薄层层析法观察,合并环黄芪醇纯度较高的流份,浓缩除去溶剂,即得纯度大于90%的环黄芪醇。
经检测本例制得的环黄芪醇,其纯度为95%。
实施例14:
A、氧化
取10g黄芪皂苷作为原料,往其中加入30%乙醇水溶液配置成黄芪皂苷含量为30g/L的悬浊液,加乙酸调节悬浊液的pH至4-5,向悬浊液中加入氧化剂——高碘酸钾在35℃下避光反应12小时,然后加入乙二醇终止氧化,其中黄芪皂苷与氧化剂的质量比为1:5;随后加氨水调pH至7-8,减压浓缩至无醇味,过滤反应液得到滤饼,用水润洗滤饼几次后,得到氧化产物。
B、还原
将A步得到的氧化产物加入50%乙醇水溶液分散成悬浊液,加入的50%乙醇水溶液与A步的黄芪皂苷的质量比为1:2,此时溶液为悬浊液;然后加入硼氢化钠作为还原剂,还原反应24h后,加入70%乙酸水溶液至反应液pH到4-5终止反应;其中加入的还原剂与A步的黄芪皂苷的质量比为1:15。
C、水解
向步骤B的反应液加入硝酸,使加入的硝酸与反应液的质量比为1:100,在10℃下搅拌反应12h。
D、萃取;
向步骤C得到的反应液中加NaOH,调pH至中性,再采用与反应液等体积乙酸乙酯萃取3次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用反相硅胶柱层析纯化:洗脱剂选用甲醇:水==50:50~100:0,通过薄层层析法观察,合并环黄芪醇纯度较高的流份,浓缩除去溶剂;再即得纯度大于90%的环黄芪醇。
经检测本例制得的环黄芪醇,其纯度为95%。
实施例15:
A、氧化
取20g黄芪皂苷作为原料,往其中加入体积比1:2的甲醇乙醇溶液配置成黄芪皂苷含量为0.6g/L的悬浊液,加磷酸调节悬浊液的pH至5-6,向悬浊液中加入氧化剂——高碘酸钾在5℃下避光反应96小时,然后加入乙二醇终止氧化,其中黄芪皂苷与氧化剂的质量比为1:2;随后加Na2CO3调pH至8-9,减压浓缩至无醇味,加入等体积正丁醇萃取4次,合并正丁醇萃取部分,水洗2次,取正丁醇部分,蒸去正丁醇,得到氧化产物。
B、还原
将A步得到的氧化产物加入加入10%乙醇水溶液分散成悬浊液,加入的10%乙醇水溶液与A步的黄芪皂苷的质量比为1:0.6;然后加入硼氢化钠作为还原剂,还原反应24h后,加入乙酸至反应液pH到4-5终止反应;其中加入的还原剂与A步的黄芪皂苷的质量比为1:15。
C、水解
向步骤B的反应液加入盐酸,使加入的盐酸与反应液的质量比为3:100,在40℃下搅拌反应96h。
D、萃取;
向步骤C得到的反应液中加NaOH,调pH至中性,再采用与反应液等体积乙酸乙酯萃取53次,合并乙酸乙酯萃取液,减压浓缩至干,得环黄芪醇粗品。
E、纯化
将步骤D得到的环黄芪醇粗品使用反相硅胶柱层析纯化:洗脱剂选用乙腈:水=30:70~80:20,通过薄层层析法观察,合并环黄芪醇纯度较高的流份,浓缩除去溶剂,即得纯度大于90%的环黄芪醇。
经检测本例制得的环黄芪醇,其纯度为96%。
药效实验一:(环黄芪醇对化疗药物的增效作用)
1、实验材料
1.1药物及试剂:环黄芪醇,由实施例13所述制备方法制得,经过检测,纯度为95%,临用前用0.5%CMC-Na溶液配成母液10mg/ml,除菌备用。5-氟尿嘧啶(5-FU)针剂,批号:20130412,,10ml/支,上海旭东海普药业有限公司。IL-2和TNF-α放免试剂盒,北京东亚放射免疫研究所。
1.2动物及瘤株
昆明种小鼠,6~8周龄,体重18~22g,购自四川大学实验动物中心,合格证号:SCXK(川)2012-0010。雌雄兼用,每批实验采用同一性别。
1.3细胞株:小鼠肝癌H22,小鼠肉瘤S180,购自四川大学华西基础医学与法医学院药理教研室。
1.4仪器与器械
无菌室和超净工作台;
血象分析仪;
外科手术器械;
消毒用碘酒、酒精。
2、试验内容
荷瘤鼠模型的建立:小鼠肝癌H22、肉瘤S180瘤株,腹腔传代接种。取于昆明鼠腹腔内接种七天的上述荷瘤小鼠,消毒腹部皮肤后,抽取腹水,离心后弃上清,无菌生理盐水稀释,调整细胞浓度为2×107个细胞/ml。消毒鼠腋下皮肤,每只小鼠于腋下注射瘤细胞悬液0.2ml,各瘤株分别接种90只。
接瘤后第二天随机分为如下9组并给药:
空白对照组(模型组):给予等量辅料的0.5%CMC-Na溶液
5-FU低剂量组:5-FU5mg/kg
5-FU低剂量+药物高剂量组:给予5-FU5+环黄芪醇30mg/kg
5-FU低剂量+药物中剂量组:给予5-FU5+环黄芪醇15mg/kg
5-FU低剂量+药物低剂量组:给予5-FU5+环黄芪醇5mg/kg
5-FU高剂量组:5-FU15mg/kg
5-FU高剂量+药物高剂量组:给予5-FU15+环黄芪醇30mg/kg
5-FU高剂量+药物中剂量组:给予5-FU15+环黄芪醇15mg/kg
5-FU高剂量+药物低剂量组:给予5-FU15+环黄芪醇5mg/kg
5-FU腹腔注射给药,其余均为灌胃给药,每日1次,连续给药10天。末次给药24h后颈椎脱臼处死小鼠,称体重,计算体重差(结束体重-开始体重),剥取瘤组织并称重,计算抑瘤率。
抑瘤率=(1-模型对照组平均瘤重/实验组平均瘤重)×100%;
胸腺(脾)指数=[胸腺(脾)重量/体重]×100%。
末次给药后脱颈椎处死小鼠,取瘤并称重,比较模型对照组、单用5-FU、5-FU与环黄芪醇不同剂量联合用药各组的抑瘤率,观察环黄芪醇对化疗药物的增效作用。
3、试验结果
3.1环黄芪醇对5-FU治疗小鼠H22肝癌的增效作用
以下的表1为试验得到的环黄芪醇对5-FU治疗小鼠H22肝癌的增效作用结果。表1表明,5-FU的5mg/kg剂量组和15mg/kg剂量组的对小鼠H22肝癌的抑瘤率分别为25.58%和46.98%,而加用环黄芪醇后,明显提高了5-FU的抑瘤效果。联合治疗组与相应剂量5-FU组比较有显著性差别(高剂量和中剂量组:P<0.01,低剂量组:P<0.05)。表明环黄芪醇对5-FU具有明显的增效作用。
表1:5-FU与环黄芪醇联合应用对小鼠H22肝癌生长的抑制作用
注:与空白对照组比较:*P<0.01,**P<0.05。
与相应剂量5-FU组比较:#P<0.01,##P<0.05。
3.2环黄芪醇对5-FU治疗小鼠S180肉瘤的增效作用
表2为环黄芪醇对5-FU治疗小鼠S180肉瘤的增效作用结果。表2表明:5-FU的5mg/kg剂量组和15mg/kg剂量组的对小鼠S180肉瘤的抑制率抑瘤率分别为25.61%和44.72%,而加用环黄芪醇后,明显提高了5-FU的抑瘤效果。联合治疗组与相应剂量5-FU组比较有显著性差别(高剂量和中剂量组:P<0.01,低剂量组:P<0.05)。表明环黄芪醇对5-FU具有明显的增效作用。
表2:5-FU与环黄芪醇联合应用对小鼠S180肉瘤生长的抑制作用
注:与空白对照组比较:*P<0.01,**P<0.05。
与相应剂量5-FU组比较:#P<0.01,##P<0.05。
药效实验二:(环黄芪醇的降低抗癌药物毒性作用)
1、试验材料:
同药效实验一
2、试验内容:用小鼠H22肝癌和S180肉瘤建立小鼠体内移植瘤模型,按如下组别分组及给药:
空白对照组(模型组):给予等量辅料的0.5%CMC-Na溶液
5-FU组:25mg/kg
5-FU+药物高剂量组:给予5-FU25+环黄芪醇30mg/kg
5-FU+药物中剂量组:给予5-FU25+环黄芪醇15mg/kg
5-FU+药物低剂量组:给予5-FU25+环黄芪醇5mg/kg
给药方法如前所述。比较空白对照组、单用5-FU治疗和5-FU与环黄芪醇各剂量联合应用治疗各组之间动物体重、胸腺指数、脾指数,用血象分析计数外周血白细胞,比较各组间白细胞数。
3、试验结果
表3、表4分别为环黄芪醇对5-FU在治疗小鼠H22肝癌中和S180肉瘤的减毒作用试验结果。表3、表4说明:在小鼠H22肝癌和S180肉瘤模型中,5-FU25mg/kg剂量组在抑瘤的同时,也使体重、免疫器官脏器指数及白细胞指数明显下降。与环黄芪醇合用后可明显改善由5-FU所致小鼠体重减轻、白细胞下降及免疫器官萎缩,提高小鼠胸腺及脾脏脏器指数。联合治疗组与相应剂量5-FU组比较有显著性差别(各剂量组:P<0.05)。表明环黄芪醇可明显降低由5-FU所致毒副作用。
表3:环黄芪醇对5-FU在治疗小鼠H22肝癌中的减毒作用
注:与空白对照组比较:*P<0.01,**P<0.05。
与相应剂量5-FU组比较:#P<0.01,##P<0.05。
表4环黄芪醇对5-FU在治疗小鼠S180肉瘤中的减毒作用
注:与空白对照组比较:*P<0.01,**P<0.05。
与相应剂量5-FU组比较:#P<0.01,##P<0.05。
药效实验三:(环黄芪醇对小鼠血清IL-2和TNF-α的影响)
1、试验材料:同药效实验一
2、试验内容:用小鼠H22肝癌建立小鼠体内移植瘤模型,按如下组别分组及给药:
空白对照组(模型组):给予等量辅料的0.5%CMC-Na溶液
5-FU组:25mg/kg
5-FU+药物高剂量组:给予5-FU25+环黄芪醇30mg/kg
5-FU+药物中剂量组:给予5-FU25+环黄芪醇15mg/kg
5-FU+药物低剂量组:给予5-FU25+环黄芪醇5mg/kg
给药方法如前所述。比较空白对照组、单用5-FU治疗和5-FU与环黄芪醇各剂量联合应用治疗各组之间IL-2和TNF-α的参数。
3、试验结果
表5为环黄芪醇对H22荷瘤小鼠血清IL-2和TNF-α的影响试验结果。表5说明:与空白对照组相比,H22荷瘤小鼠在注射5-FU后血清IL-2含量明显升高,而TNF-α含量显著降低(P<0.05)。而5-FU与环黄芪醇高、中剂量联合治疗组血清IL-2含量明显降低,TNF-α含量明显升高,与5-FU组有显著性差异(P<0.05)。
表5环黄芪醇对H22荷瘤小鼠血清IL-2和TNF-α的影响
注:与空白对照组比较:*P<0.01,**P<0.05。
与相应剂量5-FU组比较:#P<0.01,##P<0.05。
一种环黄芪醇的制备方法及其用途专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



















动态评分
0.0