







专利摘要
本发明公开了一种利用两步酶解法转化黄芪甲苷制备环黄芪醇的方法。具体涉及一种以黄芪甲苷为底物,利用两种不同的水解酶分别水解断裂黄芪甲苷C3位置的β‑木糖苷键和C6位置的β‑葡萄糖苷键而得到黄芪甲苷的苷元环黄芪醇,后经萃取、硅胶柱层析以及乙醇重结晶纯化得到纯度95%以上的环黄芪醇产品。本发明解决了化学法制备环黄芪醇过程中黄芪甲苷的三元环结构的破坏而导致的大量副产物的产生的问题以及克服了传统环黄芪醇制备方法中底物转化率低、步骤繁琐、污染环境等不足。而本发明的优点在于底物特异性高,底物黄芪甲苷全部转化,步骤简单,条件温和,成本低,是温和的生物制备方法,对环境无污染,适合工业化生产。
权利要求
1.一种利用两步酶解法转化黄芪甲苷制备环黄芪醇的方法,其特征在于,依次包括以下步骤:
A.酶解缓冲液的配制
准确配制0.2mol/L磷酸氢二钠溶液和0.1mol/L的柠檬酸溶液,将两种溶液按照1.06:1的体积比混合,并用盐酸调节混合溶液的pH至5.0;
B.第一步酶解
向步骤A中配制好的酶解缓冲体系中加入底物黄芪甲苷,底物中黄芪甲苷的质量百分比为10%,体系中黄芪甲苷的质量浓度为0.1g/L~1g/L;待底物完全溶解后加入水解酶1,水解酶1和黄芪甲苷的质量比为5:1;调节反应体系的pH为4.6~5.2,最后将反应体系置于温度45~55℃和搅拌转速为200r/min的条件下充分反应48~72h;
C.第二步酶解
用盐酸调节步骤B中反应结束后的体系pH至7.0~7.6,再向该体系中加入水解酶2,水解酶2和黄芪甲苷的质量比为50:1,最后将反应体系置于温度为25~35℃和搅拌转速为200r/min的条件下充分反应48~72h;
D.环黄芪醇的分离纯化
将步骤C中含有产物环黄芪醇的水解液经水饱和正丁醇萃取后真空浓缩蒸干得到萃取产品,并进一步利用硅胶柱层析分离纯化,最终再通过乙醇重结晶将产品的纯度提高到95%以上,得到环黄芪醇产品;
步骤B中所用的水解酶1为纤维素酶,水解断裂的是黄芪甲苷C
步骤B中得到酶解产物是6-O-葡萄糖-环黄芪醇;
步骤C中水解酶2为来源于Phycicoccus sp.Soil748的β-葡萄糖苷酶,其基因长度为1848bp,编码616个氨基酸,具体基因序列为SEQ ID No.1,编码的蛋白质序列为SEQ IDNo.2,水解断裂的是中间产物6-O-葡萄糖-环黄芪醇上C
步骤C中β-葡萄糖苷酶的底物为6-O-葡萄糖-环黄芪醇,步骤C中得到的产物为环黄芪醇。
2.根据权利要求1所述的方法,其特征在于:步骤C中的β-葡萄糖苷酶通过将β-葡萄糖苷酶基因导入大肠杆菌后诱导表达,再将菌体经超声破碎后而得到的粗酶液,每100mL的发酵菌液破碎后制得20mLβ-葡萄糖苷酶粗酶液。
3.根据权利要求1所述的方法,其特征在于:步骤D中的水解液和水饱和正丁醇的体积比为1:0.5,萃取次数为三遍。
4.根据权利要求1所述的方法,其特征在于:步骤D中的硅胶柱层析填料为100~200目硅胶粉。
5.根据权利要求1所述的方法,其特征在于:步骤D中硅胶柱层析的洗脱剂为甲醇-氯仿-水的下层溶液,体积比为13:4:2。
说明书
技术领域
本发明属于医药化工领域,具体涉及一种利用两步酶解法转化黄芪甲苷
制备环黄芪醇的方法。
背景技术
黄芪甲苷是中药黄芪的主要活性成分,具有提高免疫力、清除自由基、降压、保肝利尿以及抗菌等多种功效。而环黄芪醇为黄芪皂苷的苷元,研究显示其具有抗氧化、能提高免疫力以及抵抗衰老等活性作用,且相比黄芪皂苷更易于肠道吸收以及膜渗透。另有研究显示,黄芪皂苷在体内发挥药物效果主要是通过被部分分解成环黄芪醇而起到作用。最重要的是环黄芪醇是目前世界上唯一已被证明的可以激活端粒酶活性的活性分子,能够有效抑制端粒的减少。
环黄芪醇(CA):C30H50O5、490.71、环黄芪醇黄芪皂苷的苷元,主要由黄芪甲苷水解得到,为无色针状结晶,易溶于甲醇、正丁醇等。环黄芪醇的化学结构式如下:
对于环黄芪醇的发现在1983年就有报道,它是大部分黄芪皂苷的苷元。目前主要通过化学法水解断裂黄芪甲苷(ASI)C3位置的木糖苷键和C6位置的葡萄糖苷键来得到环黄芪醇,但是由于黄芪甲苷上有一个三元环结构,这种结构不稳定,化学法水解过程中容易开环,形成副产物黄芪醇。
中国已公开的专利申请中(中国专利申请公开号:CN 104817610 A)使用硫酸酸解黄芪甲苷制备环黄芪醇,其水解温度为130℃,反应器采用一种高压反应釜。硫酸酸解反应条件剧烈,会导致黄芪甲苷的三元环严重破坏,产生大量的副产物。中国已公开的专利申请中(中国专利申请公开号:CN 103880910 A)采用一种氧化还原的方法制备环黄芪醇,理论上解决了三元环开环的问题,但是其操作步骤繁琐,而且强氧化剂和强还原剂的使用增加了对环境的污染以及生产成本,不易工业化生产。
另外中国已公开的两篇专利申请中(中国专利申请公开号:CN 105734109 A、CN105566434 A)均使用了多种水解酶复合的形式水解黄芪甲苷制备环黄芪醇,其中专利CN105734109 A中的复合酶为β-葡萄糖苷酶、蜗牛酶、柚苷酶、纤维素酶等酶按不同质量比的形式混合得来,而专利CN 105566434 A中的复合酶为β-葡萄糖苷酶、柚苷酶、β-木糖苷酶的组合。相较于本发明来说,复合酶的缺点无形中提高了酶成本,而且复合酶对底物黄芪甲苷的专一性差,黄芪甲苷的转化率较低。
发明内容
本发明选择采用一种两步酶解法转化黄芪甲苷制备环黄芪醇,采用这种温和的生物酶法制备环黄芪醇的目的在于:一、解决传统化学法制备过程中三元环易开裂形成副产物黄芪醇的问题。二、克服化学法制备环黄芪醇中操作步骤繁琐、生产成本高、污染环境、转化率低等缺点。三、较之一些使用复合酶酶解黄芪甲苷制备环黄芪醇的方法,本发明提供了专一性非常高的水解酶,降低了生产成本,且本发明中的黄芪甲苷可以完全转化为环黄芪醇。本发明是根据现有技术的不足作出了创新和改进,提供一种高效的易工业化生产的生物酶法制备环黄芪醇的方法。
为实现上述发明目的,所采用的技术方案是:
A.酶解缓冲液的配制
准确配制0.2mol/L磷酸氢二钠溶液和0.1mol/L的柠檬酸溶液,将两种溶液按照1.06:1的体积比混合,并用盐酸调节混合溶液的pH至5.0。
B.第一步酶解
向步骤A中配制好的酶解缓冲体系中加入底物黄芪甲苷,底物中黄芪甲苷的质量百分比为10%,体系中黄芪甲苷的质量浓度为0.1g/L~1g/L。待底物完全溶解后加入水解酶1,水解酶1和黄芪甲苷的质量比为5:1。调节反应体系的pH为4.6~5.2,最后将反应体系置于温度45~55℃和搅拌转速为200r/min的条件下充分反应48~72h;
C.第二步酶解
用盐酸调节步骤B中反应结束后的体系pH至7.0~7.6,再向该体系中加入水解酶2,水解酶2和黄芪甲苷的质量比为50:1,最后将反应体系置于温度为25~35℃和搅拌转速为200r/min的条件下充分反应48~72h。
D.环黄芪醇的分离纯化
将步骤C中含有产物环黄芪醇的水解液经水饱和正丁醇萃取后真空浓缩蒸干得到萃取产品,并进一步利用硅胶柱层析分离纯化,最终再通过乙醇重结晶将产品的纯度提高到95%以上,得到环黄芪醇产品。
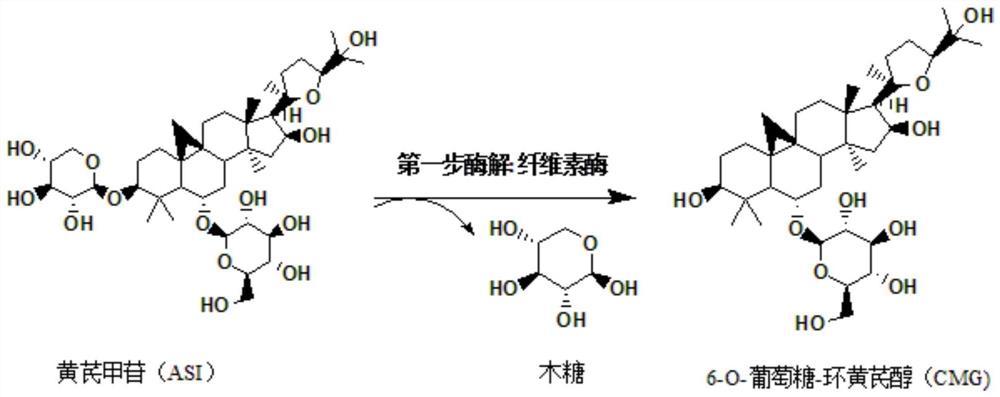
上述步骤B中所用的水解酶1为纤维素酶,水解断裂的是黄芪甲苷C3位置的木糖糖苷键,得到酶解产物是6-O-葡萄糖-环黄芪醇。
上述步骤C中水解酶2为β-葡萄糖苷酶,水解断裂的是中间产物6-O-葡萄糖-环黄芪醇上C6位置的葡萄糖苷键。β-葡萄糖苷酶的底物为步骤B中的产物,步骤C的产物为环黄芪醇。
上述步骤C中的β-葡萄糖苷酶的基因来源于Phycicoccus sp.Soil748,其基因长度为1848bp,编码616个氨基酸,具体基因序列为SEQ ID No.1,编码的蛋白质序列为SEQ IDNo.2。
上述步骤C中使用的β-葡萄糖苷酶是自制得来,具体是通过将β-葡萄糖苷酶基因导入大肠杆菌后诱导表达,再将菌体经超声破碎后而得到的粗酶液。每100mL的发酵菌液破碎后制得20mLβ-葡萄糖苷酶粗酶液。
上述步骤D中的酶解液和萃取剂的体积比为1:0.5,萃取次数为三遍。
上述步骤D中的硅胶柱层析填料为100~200目硅胶粉。
上述步骤D中硅胶柱层析的洗脱剂为甲醇-氯仿-水的下层溶液,体积比为13:4:2。
本发明的特点是利用一种两步酶法转化黄芪甲苷制备环黄芪醇。与现有技术相比,本发明显著的有益效果是:
与传统的化学法制备环黄芪醇相比,酶解法解决了传统化学法制备过程中黄芪甲苷三元环易开裂形成副产物黄芪醇的问题,本发明的酶解过程中没有副产物黄芪醇产生。
本发明中的纤维素酶和β-葡萄糖苷酶对黄芪甲苷的专一性更好,且两步酶解法能将黄芪甲苷完全转化为环黄芪醇,转化效率大幅提高。
与传统技术相比,两步酶解转化法是一种温和的生物制备方法,制备条件温和,无需高温高压,大大降低了能耗;
酶法制备无需强氧化剂、还原剂以及强酸等污染环境的试剂,是一种环境保护性的环黄芪醇制备方法。
酶法操作步骤简洁,其中的β-葡萄糖苷酶可以自制,生产成本大大降低,且最终环黄芪醇产品的纯度高,适合工业化生产。
附图说明
图1为纤维素酶转化黄芪甲苷路径图。
图2为β-葡萄糖苷酶转化6-O-葡萄糖-环黄芪醇路径图。
具体实施方式
下面结合具体实施例对本发明作进一步的描述,但这些实施例的目的并不在于限制本发明的保护范围。
实施例1
准确配制0.2mol/L磷酸氢二钠溶液和0.1mol/L的柠檬酸溶液,将两种溶液按照1.06:1的体积比混合,并用盐酸调节缓冲盐溶液的pH至5.0。
第一步酶解:准确量取100mL的酶解缓冲盐溶液置于250mL的烧杯中,并向其中加入100mg质量百分比为10%的黄芪甲苷,待底物黄芪甲苷完全溶解后将体系加热至45℃,后向体系中加入0.05g已经加热至45℃的纤维素酶,并用盐酸将体系的pH调节至4.6,最后将反应体系置于磁力加热搅拌仪器上,在搅拌转速为200r/min,温度为45℃下充分反应60h。后取样1mL过0.22μm滤膜,于高效液相色谱检测,结果显示底物黄芪甲苷完全转化为中间产物6-O-葡萄糖-环黄芪醇。
第二步酶解:在第一步酶解结束之后,用盐酸调节第一步反应的pH至7.0,并将体系温度降至25℃,再向此体系中加入0.5g已加热至25℃的β-葡萄糖苷酶粗酶液,粗酶液的获得具体是通过将β-葡萄糖苷酶基因导入大肠杆菌后诱导表达,再将菌体经超声破碎后而得到的粗酶液。每100mL的发酵菌液破碎后制得20mLβ-葡萄糖苷酶粗酶液。最后将反应体系置于磁力加热搅拌仪器上,在搅拌转速为200r/min,温度为25℃下充分反应60h。后取样1mL过0.22μm滤膜,于高效液相色谱检测,结果显示中间产物6-O-葡萄糖-环黄芪醇完全转化为终产物环黄芪醇。并收集得到92mL的酶水解液。
向两步酶解反应结束后的92mL含有产物环黄芪醇的水解液中加入50mL水饱和正丁醇萃取,一共萃取三次,合并三次的正丁醇相并真空浓缩蒸干得到萃取产品79.68mg,后用10mL的甲醇将萃取产品完全溶解,取1mL溶液过0.22μm有机滤膜,于高效液相色谱-蒸发光检测下测定,经计算萃取产品的纯度为7.05%。
采用上述方法制取纯度为7.05%的粗产品1g并于硅胶柱层析分离纯化,硅胶柱填料为100~200目的硅胶粉,采用干法上样,洗脱剂为甲醇-氯仿-水的下层溶液,其比例为13:4:2。收集其中含有产品的馏分并合并,后于液相检测浓度。最后将所有的馏分收集旋干并称量干重,经计算得到79.43mg纯度为84.36%硅胶层析产品。最后将硅胶柱层析产品通过乙醇重结晶将产品的纯度提高到95%以上,得到环黄芪醇产品。
实施例2
准确配制0.2mol/L磷酸氢二钠溶液和0.1mol/L的柠檬酸溶液,将两种溶液按照1.06:1的体积比混合,并用盐酸调节缓冲盐溶液的pH至5.0。
第一步酶解:准确量取100mL的酶解缓冲盐溶液置于250mL的烧杯中,并向其中加入100mg质量百分比为10%的黄芪甲苷,待底物黄芪甲苷完全溶解后将体系加热至50℃,后向体系中加入0.05g已经加热至50℃的纤维素酶,并用盐酸将体系的pH调节至5.0,最后将反应体系置于磁力加热搅拌仪器上,在搅拌转速为200r/min,温度为50℃下充分反应48h。后取样1mL过0.22μm滤膜,于高效液相色谱检测,结果显示底物黄芪甲苷完全转化为中间产物6-O-葡萄糖-环黄芪醇。
第二步酶解:在第一步酶解结束之后,用盐酸调节第一步反应的pH至7.4,并将体系温度降至30℃,再向此体系中加入0.5g已加热至30℃的β-葡萄糖苷酶粗酶液,粗酶液的获得具体是通过将β-葡萄糖苷酶基因导入大肠杆菌后诱导表达,再将菌体经超声破碎后而得到的粗酶液。每100mL的发酵菌液破碎后制得20mLβ-葡萄糖苷酶粗酶液。最后将反应体系置于磁力加热搅拌仪器上,在搅拌转速为200r/min,温度为30℃下充分反应48h。后取样1mL过0.22μm滤膜,于高效液相色谱检测,结果显示中间产物6-O-葡萄糖-环黄芪醇完全转化为终产物环黄芪醇。并收集得到94mL的酶水解液。
向两步酶解反应结束后的94mL含有产物环黄芪醇的水解液中加入50mL水饱和正丁醇萃取,一共萃取三次,合并三次的正丁醇相并真空浓缩蒸干得到萃取产品75.35mg,后用10mL的甲醇将萃取产品完全溶解,取1mL溶液过0.22μm有机滤膜,于高效液相色谱-蒸发光检测下测定,经计算萃取产品的纯度为7.35%。
采用上述方法制取纯度为7.35%的粗产品1g并于硅胶柱层析分离纯化,硅胶柱填料为100~200目的硅胶粉,采用干法上样,洗脱剂为甲醇-氯仿-水的下层溶液,其比例为13:4:2。收集其中含有产品的馏分并合并,后于液相检测浓度。最后将所有的馏分收集旋干并称量干重,经计算得到81.98mg纯度为85.17%硅胶层析产品。最后将硅胶柱层析产品通过乙醇重结晶将产品的纯度提高到95%以上,得到环黄芪醇产品。
实施例3
准确配制0.2mol/L磷酸氢二钠溶液和0.1mol/L的柠檬酸溶液,将两种溶液按照1.06:1的体积比混合,并用盐酸调节缓冲盐溶液的pH至5.0。
第一步酶解:准确量取100mL的酶解缓冲盐溶液置于250mL的烧杯中,并向其中加入100mg质量百分比为10%的黄芪甲苷,待底物黄芪甲苷完全溶解后将体系加热至55℃,后向体系中加入0.05g已经加热至55℃的纤维素酶,并用盐酸将体系的pH调节至5.5,最后将反应体系置于磁力加热搅拌仪器上,在搅拌转速为200r/min,温度为55℃下充分反应72h。后取样1mL过0.22μm滤膜,于高效液相色谱检测,结果显示底物黄芪甲苷完全转化为中间产物6-O-葡萄糖-环黄芪醇。
第二步酶解:在第一步酶解结束之后,用盐酸调节第一步反应的pH至7.6,并将体系温度降至35℃,再向此体系中加入0.5g已加热至35℃的β-葡萄糖苷酶粗酶液,粗酶液的获得具体是通过将β-葡萄糖苷酶基因导入大肠杆菌后诱导表达,再将菌体经超声破碎后而得到的粗酶液。每100mL的发酵菌液破碎后制得20mLβ-葡萄糖苷酶粗酶液。最后将反应体系置于磁力加热搅拌仪器上,在搅拌转速为200r/min,温度为35℃下充分反应72h。后取样1mL过0.22μm滤膜,于高效液相色谱检测,结果显示中间产物6-O-葡萄糖-环黄芪醇完全转化为终产物环黄芪醇。并收集得到93mL的酶水解液。
向两步酶解反应结束后的93mL含有产物环黄芪醇的水解液中加入50mL水饱和正丁醇萃取,一共萃取三次,合并三次的正丁醇相并真空浓缩蒸干得到萃取产品78.38mg,后用10mL的甲醇将萃取产品完全溶解,取1mL溶液过0.22μm有机滤膜,于高效液相色谱-蒸发光检测下测定,经计算萃取产品的纯度为6.55%。
采用上述方法制取纯度为6.55%的粗产品1g并于硅胶柱层析分离纯化,硅胶柱填料为100~200目的硅胶粉,采用干法上样,洗脱剂为甲醇-氯仿-水的下层溶液,其比例为13:4:2。收集其中含有产品的馏分并合并,后于液相检测浓度。最后将所有的馏分收集旋干并称量干重,经计算得到79.44mg纯度为82.46%硅胶层析产品。最后将硅胶柱层析产品通过乙醇重结晶将产品的纯度提高到95%以上,得到环黄芪醇产品。
实施例4
准确配制0.2mol/L磷酸氢二钠溶液和0.1mol/L的柠檬酸溶液,将两种溶液按照1.06:1的体积比混合,并用盐酸调节缓冲盐溶液的pH至5.0。
第一步酶解:准确量取100mL的酶解缓冲盐溶液置于250mL的烧杯中,并向其中加入1000mg质量百分比为10%的黄芪甲苷,待底物黄芪甲苷完全溶解后将体系加热至50℃,后向体系中加入0.5g已经加热至50℃的纤维素酶,并用盐酸将体系的pH调节至5.0,最后将反应体系置于磁力加热搅拌仪器上,在搅拌转速为200r/min,温度为50℃下充分反应48h。后取样1mL过0.22μm滤膜,于高效液相色谱检测,结果显示底物黄芪甲苷完全转化为中间产物6-O-葡萄糖-环黄芪醇。
第二步酶解:在第一步酶解结束之后,用盐酸调节第一步反应的pH至7.4,并将体系温度降至30℃,再向此体系中加入5g已加热至30℃的β-葡萄糖苷酶粗酶液,粗酶液的获得具体是通过将β-葡萄糖苷酶基因导入大肠杆菌后诱导表达,再将菌体经超声破碎后而得到的粗酶液。每100mL的发酵菌液破碎后制得20mLβ-葡萄糖苷酶粗酶液。最后将反应体系置于磁力加热搅拌仪器上,在搅拌转速为200r/min,温度为30℃下充分反应48h。后取样1mL过0.22μm滤膜,于高效液相色谱检测,结果显示中间产物6-O-葡萄糖-环黄芪醇完全转化为终产物环黄芪醇。并收集得到990mL的酶水解液。
向两步酶解反应结束后的990mL含有产物环黄芪醇的水解液中加入50mL水饱和正丁醇萃取,一共萃取三次,合并三次的正丁醇相并真空浓缩蒸干得到萃取产品770.28mg,取100mg萃取产品用10mL的甲醇完全溶解,取1mL溶液过0.22μm有机滤膜,于高效液相色谱-蒸发光检测下测定,经计算萃取产品的纯度为7.26%。
采用上述方法制取纯度为7.26%的粗产品10g并于硅胶柱层析分离纯化,硅胶柱填料为100~200目的硅胶粉,采用干法上样,洗脱剂为甲醇-氯仿-水的下层溶液,其比例为13:4:2。收集其中含有产品的馏分并合并,后于液相检测浓度。最后将所有的馏分收集旋干并称量干重,经计算得到891.23mg纯度为81.46%硅胶层析产品。最后将硅胶柱层析产品通过乙醇重结晶将产品的纯度提高到95%以上,得到环黄芪醇产品。
序列表
<110> 北京化工大学
<120> 一种利用两步酶解法转化黄芪甲苷制备环黄芪醇的方法
<140> 2017103197960
<141> 2017-05-09
<160> 2
<170> SIPOSequenceListing 1.0
<210> 1
<211> 1848
<212> DNA
<213> Phycicoccus sp. Soil748
<400> 1
atgcagccgg agcgtcagac ttccccggaa ggtgtagctt accgtgacct gaacggtaac 60
ggtcgtatgg acccgtacga ggacccgcgt ctgcctgtag aggaacgtgt agaagacctg 120
ctgggtcgtc tgtctctgga ggagaaggca ggtctgatgt tccacaccgt gatcgaggct 180
ggcaccgacg gtacggtact ggagcaccca ggtgcaatca gcaagtcccc taccagcgag 240
gtcgtcctgt ctaagcacct gacccacttc aacgtgcatg ccctggacga cgctcgtatg 300
gctgcacgct ggcataacgc actgcaagca ctggctgagc agactccgca tggtatcccg 360
gtaaccgtct ccactgatcc acgtcacgct ttcatcgaga acgcgggtgt gagcttcacc 420
gcgaaagcat tctcccagtg gccggaacca ctgggtctgg cagcactgcg tgacgcagag 480
gcagtccgtg cattcggtga catcgcacgt aaagaatacc gtgcggtcgg tatccgtgcg 540
gctctgcacc caactctgga cctggcaact gaaccacgtt gggcacgtca ggctggtacg 600
ttcggtcaag accctgacct ggtgacggaa ctgggtgtcg cttacctgaa aggcttccag 660
ggcgaagcgc tgggtccgga tagcgtagca tgtaccagca aacacttccc aggcggtggt 720
ccgcagaaag acggtgaaga cgctcacttc ccgtatggcc gtgaacaggt gtacccgggt 780
gaccgtttcg gtgatcatct gaaaccgttc ccgccgatta tcgaagccgg caccgccgct 840
atcatgccgt actatggcat gcctgttggt ctggtgctgg gcggcgaacc gatcgaagaa 900
gtgggcttcg gctacaaccg ccagattgtg accggcctgc tgcgcgaaca gctgggttac 960
gacggtgttg tcgtgaccga ctgggaactg gtgaatgaca atcacgtggg cgaccaggtt 1020
ctgccagccc gtgcatgggg tgttgaacac ctggacccgc acggccgtat ggaactgatc 1080
ctgcacgccg gtgctgatca gtttggtggt gaagaatgcg tggaaattct gctggagctg 1140
gtgaaagaag gccgtgtgac cgaagaacgt attgacgaaa gcgcgcgccg tctgctggcc 1200
gttaaattcc gtctgggcct gttcgacgat ccgtacgttg atgaagatct ggccgctacc 1260
accgtgggtc gtgaagattt ccgcgaagcg ggctatgcgg cccaggcccg ctccgtaacc 1320
gttctgcaag atggctctgg tgatctggcg cctgttctgc cgctggcccg cggccgtaaa 1380
gtttatgcgg aaaacgtttc cgatgaagct ctggcggcgg ttggcacgcg tgtagcgact 1440
ccggaagaag cggatgttgc gctggttcgc ctgatggctc cgttcgaacc gcgcagcgac 1500
ctgttcctgg aatcctggtt tcatcagggc tctctggatt ttccgcctgg cctggttgcg 1560
cgcctggctc gtgttgccgc tcactgcccg ctggttgttg atgttgttct ggatcgcccg 1620
gctgttctga ccccgctgct gacttttgct tctgcggtag ttggctctta tggcacctct 1680
gatgcggcgc tgctggatgc gctgactggc actattccgc cggtaggccg cctgccgttt 1740
gatctgccgc gctctatgga agatgtacgc cgcaaaggtg aagatgttcc gggctttgcg 1800
gacccgctgt ttccgtttgg tcatggcctg gcgctgcgtg atccgtct 1848
<210> 2
<211> 396
<212> DNA/RNA
<213> Phycicoccus sp. Soil748
<400> 2
mrtsgvayrd ngngrmdydr vrvdgrskag mhtvagtdgt vhgaskstsv vskhthnvha 60
ddarmaarwh naaathgvtv stdrhanagv stakaswgaa rdaavragda rkyravgraa 120
htdatrwara gtgddvtgva ykggagdsva ctskhgggkd gdahygrvyg drgdhkagta 180
amyygmvgvg gvggynrvtg rgydgvvvtd wvndnhvgdv arawgvhdhg rmhagadggc 240
vvkgrvtrds arravkrgdd yvddaattvg rdragyaaar svtvdgsgda vargrkvyan 300
vsdaaavgtr vatadvavrm arsdswhgsd gvararvaah cvvdvvdrav ttasavvgsy 360
gtsdaadatg tvgrdrsmdv rrkgdvgadg hgards 396
一种利用两步酶解法转化黄芪甲苷制备环黄芪醇的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。


















动态评分
0.0