








专利摘要
本申请公开了一种合成钙钛矿纳米晶的方法,所述方法至少包括以下步骤:S1)获得含有有机钛酯的体系I;S2)在搅拌条件下,将钙钛矿前驱体加入步骤S1)中所述体系I中反应,得到所述钙钛矿纳米晶材料。所述方法能够实现原料直接在室温下混合,快捷简单,便于操作,有望广泛应用于大规模铅卤钙钛矿型量子点的合成。
权利要求
1.一种合成钙钛矿纳米晶的方法,其特征在于,至少包括以下步骤:
S1)获得含有有机钛酯的体系I;
S2)在搅拌条件下,将钙钛矿前驱体加入步骤S1)中所述体系I中反应,得到所述钙钛矿纳米晶材料。
2.根据权利要求1所述的方法,其特征在于,步骤S1)和步骤S2)均在室温、空气条件下进行。
3.根据权利要求1所述的方法,其特征在于,步骤S1)中所述有机钛酯选自钛酸异丙酯、钛酸四乙酯、钛酸正四丁酯、钛酸四叔丁酯中的至少一种。
4.根据权利要求1所述的方法,其特征在于,步骤(S1)中所述体系I中还包括溶剂和表面活性剂;
优选地,所述溶剂选自具有式(I)所示化学式的化合物中的至少一种:
R
其中,R
优选地,所述表面活性剂选自具有式(II)、式(III)中所示化学式的化合物中的至少一种:
R
R
其中,R
优选地,所述溶剂为十八烯;所述表面活性剂选自油酸、油胺中的至少一种;
优选地,所述溶剂、表面活性剂和有机钛酯的体积比例满足:
溶剂:表面活性剂:有机钛酯=8~12:0.5~2:0.5~2;
优选地,所述溶剂、表面活性剂和有机钛酯的体积比例满足:
溶剂:表面活性剂:有机钛酯=8~12:1~2:0.5~2;
优选地,所述R
优选地,所述R
5.根据权利要求1所述的方法,其特征在于,步骤S2)中所述钙钛矿选自具有式(IV)所示化学式中的至少一种:
AMX
其中,A选自CH
M为金属离子,所述金属选自Sn、Pb中的至少一种;
所述X选自卤素阴离子中的至少一种;
优选地,所述钙钛矿纳米晶为CsPbBr
优选地,所述步骤S2)中钙钛矿前驱体包括盐类化合物AX
所述X
优选地,所述X
6.根据权利要求5所述的方法,其特征在于,步骤S2)中所述AX
优选地,步骤S2)中所述AX
优选地,步骤S2)中所述AX
优选地,步骤S2)中所述AX
优选地,步骤S2)中所述反应条件选自机械搅拌、磁力搅拌、高速分散、振荡中的至少一种;
其中,所述搅拌的速率为400~640rpm/min。
7.根据权利要求1所述的方法,其特征在于,步骤S2)中所述反应时间为5~240分钟;
优选地,步骤S2)中所述反应时间为30~180分钟。
8.根据权利要求1所述的方法,其特征在于,所述方法至少包括:
步骤a1:将表面活性剂加入到溶剂中,然后加入有机钛酯,搅拌;
步骤a2:将钙钛矿前驱体加入到步骤a1中搅拌的溶液中反应;
步骤a3:将步骤a2中反应后的溶液离心,保留下层沉淀,即得到所述钙钛矿纳米晶;
优选地,所述方法至少包括:
步骤b1:将10ml十八烯加入到20ml的试剂瓶中,加入0.5-1ml的油酸和0.5-1ml的油胺,然后加入0.5-2ml的有机钛酯,不断搅拌;
步骤b2:称量0.1-0.4mmol的PbBr
步骤b3:将反应后的溶液在11000rpm条件下离心5分钟,保留下层沉淀中的CsPbBr
9.根据权利要求1所述的方法,其特征在于,所述钙钛矿纳米晶材料的尺寸为10~100nm;
优选地,所述钙钛矿纳米晶材料的尺寸为15~50nm;
优选地,所述钙钛矿纳米晶材料的发光波长在480~520nm。
10.一种发光材料,其特征在于,包含根据权利要求1至9任一项所述方法制备得到的钙钛矿纳米晶材料。
说明书
技术领域
本申请涉及一种钙钛矿纳米晶材料的制备方法,属于材料领域。
背景技术
铅卤钙钛矿型纳米晶体,由于其优异的光电性能成为近年来研究的热点。其光致发光产率在无任何附加表面钝化的条件下可达到90%。光学特性在整个可见光谱范围内可以调节。这些光学特性使其在各种光电子应用领域作为新型材料而崭露头角,例如用于可发射激光的高效LED的低阈值单光子和双光子泵浦增益材料。
传统的铅卤钙钛矿型量子点的制备通常采用热注入法。但是热注入法需要较为苛刻和繁琐的合成条件,如高温、惰性气体保护和局部注射操作。这些大大增加了它在实际应用中的局限性。对于合成方法的改进是目前关于铅卤钙钛矿的研究重点。
发明内容
根据本申请的一个方面,提供了一种钙钛矿纳米晶的制备的方法,该方法能够实现原料直接在室温下混合,快捷简单,便于操作,有望广泛应用于大规模铅卤钙钛矿型量子点的合成。
所述钙钛矿纳米晶的制备方法,其特征在于,至少包括以下步骤:
S1)获得含有有机钛酯的体系I;
S2)在搅拌条件下,将钙钛矿前驱体加入步骤S1)中所述体系I中反应,得到所述钙钛矿纳米晶材料。
所述方法适用于常见的钙钛矿纳米晶的制备,优选AMX3型钙钛矿纳米微晶,优选铅卤钙钛矿型量子点;优选所述钙钛矿材料为CsPbX3;进一步优选地,所述钙钛矿材料为CsPbBr3。其中,所述A选自CH3NH3
可选地,所述钙钛矿纳米晶材料的尺寸为10~100nm。
可选地,所述钙钛矿纳米晶材料的尺寸为10~80nm。
可选地,所述钙钛矿纳米晶材料的尺寸为15~50nm。
可选地,所述钙钛矿纳米晶材料的尺寸为15~25nm。
可选地,所述钙钛矿纳米晶材料的尺寸上限选自50nm、60nm、80nm、90nm或100nm;下限选自10nm、15nm、20nm、25nm或40nm。
可选地,所述钙钛矿纳米晶材料选自纳米片、纳米立方块、纳米棒中的至少一种。
可选地,所述纳米片的厚度为3~7nm。
可选地,所述纳米片的厚度为5nm。
可选地,所述钙钛矿纳米晶材料的发光波长在480~520nm。
可选地,所述钙钛矿纳米晶材料的发光波长上限选自490nm、495nm、500nm、509nm、510nm或520nm;下限选自480nm、485nm或490nm。
可选地,步骤S1)和步骤S2)的反应条件均在室温、空气条件下进行。
可选地,步骤S1)中所述有机钛酯选自钛酸异丙酯、钛酸四乙酯、钛酸正四丁酯、钛酸四叔丁酯中的至少一种。
可选地,所述有机钛酯选自钛酸异丙酯、钛酸四乙酯、钛酸正四丁酯中的至少一种。
可选地,步骤S1)中所述体系I还包括溶剂和表面活性剂。
可选地,所述溶剂选自具有式(I)所示化学式的化合物中的至少一种:
R
其中,R
可选地,所述表面活性剂选自具有式(II)、式(III)中所示化学式的化合物中的至少一种:
R
R
其中,R
可选地,所述溶剂为十八烯,所述表面活性剂选自油酸、油胺中的至少一种。
可选地,所述溶剂、表面活性剂和有机钛酯的体积比例满足:
溶剂:表面活性剂:有机钛酯=8~12:0.5~2:0.5~2。
可选地,所述溶剂、表面活性剂和有机钛酯的体积比例满足:
溶剂:表面活性剂:有机钛酯=8~12:1~2:0.5~2。
可选地,所述R
可选地,上述比例范围(R
可选地,所述R
可选地,步骤S2)中所述钙钛矿选自具有式(IV)所示化学式中的至少一种:
AMX3 式(IV)
其中,A选自CH3NH3
M为金属离子,所述金属选自Sn、Pb中的至少一种;
所述X选自卤素阴离子中的至少一种。
可选地,所述钙钛矿纳米晶为CsPbBr3纳米晶。
可选地,所述钙钛矿前驱体包括盐类化合物AX
所述X
可选地,所述X
可选地,AX
可选地,AX
可选地,MX
可选地,所述X
可选地,步骤S2)中所述AX
可选地,步骤S2)中所述AX
可选地,步骤S2)中所述AX
可选地,步骤S2)中所述AX
可选地,步骤S2)中所述反应条件选自机械搅拌、磁力搅拌、高速分散、振荡中的至少一种。
可选地,所述搅拌的速率为400~640rpm/min。
可选地,所述振荡频率为560每分钟。
可选地,步骤S2)中所述反应时间为5~240分钟。
可选地,所述反应时间的上限选自180分钟、200分钟、220分钟、240分钟;下限选自5分钟、15分钟、30分钟、60分钟、80分钟、100分钟、120分钟、140分钟、160分钟。
可选地,步骤S2)中所述反应时间为30~180分钟。
可选地,所述反应时间的上限选自80分钟、100分钟、120分钟、140分钟、160分钟、180分钟;下限选自30分钟、60分钟、80分钟、100分钟、120分钟、140分钟、160分钟。
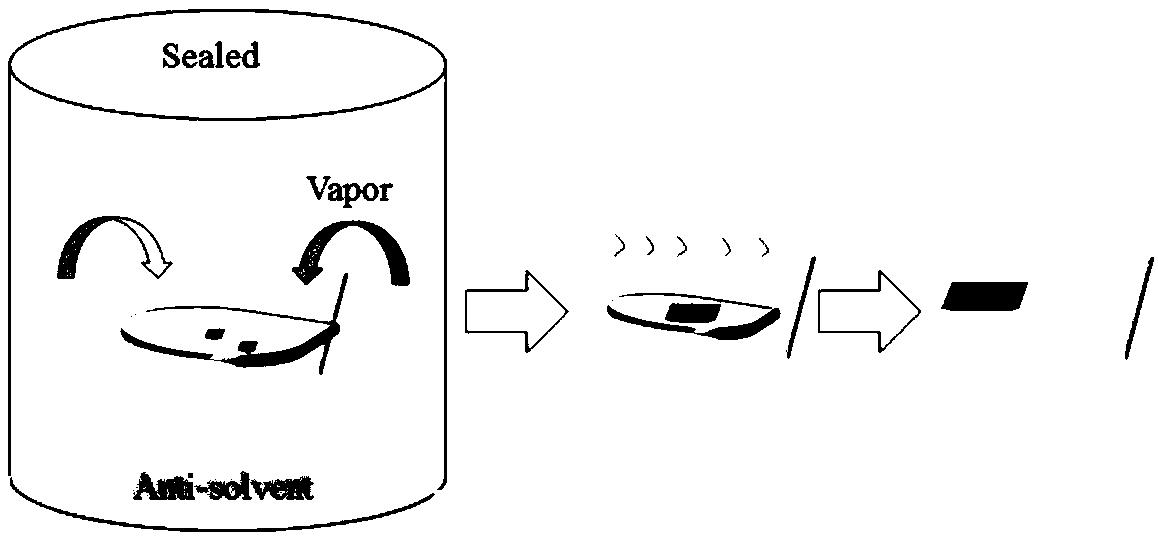
可选地,所述步骤S2)为将钙钛矿前驱体加入步骤S1)中所述体系I中反应,反应液分离得到沉淀,并用甲苯分散得到所述钙钛矿纳米晶材料。
可选地,所述分离为11000rpm离心5分钟。
可选地,所述方法至少包括:
步骤a1:将表面活性剂加入到溶剂中,然后加入有机钛酯,搅拌;
步骤a2:将钙钛矿前驱体加入到步骤a1中搅拌的溶液中反应;
步骤a3:将步骤a2中反应后的溶液离心,保留下层沉淀,即得到所述钙钛矿纳米晶;
可选地,所述方法至少包括:
步骤c1:将10ml R
步骤c2:称量0.1-0.4mmol的MX
步骤c3:将反应后的溶液在11000rpm条件下离心5分钟,保留下层沉淀中的纳米晶并用甲苯分散保存,即得到所述钙钛矿纳米晶。
可选地,所述方法至少包括:
步骤b1:将10ml十八烯加入到20ml的试剂瓶中,加入0.5-1ml的油酸和0.5-1ml的油胺,然后加入0.5-2ml的有机钛酯,不断搅拌;
步骤b2:称量0.1-0.4mmol的PbBr2和0.1mmol的Cs2CO3加入到上述不断搅拌的溶液中反应;
步骤b3:将反应后的溶液在11000rpm条件下离心5分钟,保留下层沉淀中的CsPbBr3纳米晶并用甲苯分散保存,即得到所述钙钛矿纳米晶。
本申请中将反应物扩大20倍后,该方法仍然有效,仍能合成出钙钛矿纳米晶(优选CsPbX3纳米晶)。
本申请中将反应物扩大20倍后,该方法仍然有效,仍能合成出CsPbX3纳米晶。
本申请提供了一种CsPbBr3纳米晶、其制备方法及其在荧光材料中的应用。所述CsPbBr3纳米晶可通过有机钛酯的催化快速合成。该方法只需一步操作,并可以大量合成。通过改变有机钛酯的种类可以改变纳米晶的形貌,如纳米片、纳米立方块等。该CsPbBr3纳米晶可用于LED等光电器件,在LED领域具有广阔的应用前景。
具体地,本发明目的之一在于针对目前CsPbX3(X=Cl、Br、I)纳米晶材料体系中比较缺乏的快速合成,提供一种CsPbBr3纳米晶的快速制备方法,该方法直接将固体反应物和试剂直接混合,然后在有机钛酯的催化作用下,经过室温搅拌直接合成得到了发光性能优异的钙钛矿纳米晶,这是目前合成CsPbX3纳米晶最快最简便的方法。而且将反应物扩大20倍后,该方法仍然有效,仍能合成出CsPbX3纳米晶。有机钛酯起到了催化剂的作用,并且改变有机钛酯的种类可以得到不同形貌的CsPbX3纳米晶。所述的材料发光光谱覆盖范围大,量子效率高,易于规模化合成,在白光LED照明和显示、光探测器、太阳能电池等领域具有广阔的应用前景。
具体地,所述方法至少包括以下步骤:
步骤1:将10ml十八烯加入到20ml的试剂瓶中,加入0.5-1ml的油酸和0.5-1ml的油胺,然后加入0.5-2ml的有机钛酯,不断搅拌;
步骤2:称量0.1-0.4mmol的PbBr2和0.1mmol的Cs2CO3加入到上述不断搅拌的溶液中,在室温及空气环境中,几分钟后即可观察到溶液颜色变为黄色,紫外灯(365nm)照射下可观察到明亮的绿色光;
步骤3:将反应后的溶液在11000rpm条件下离心5分钟,保留下层沉淀中的CsPbBr3纳米晶并用甲苯分散保存。
具体地,反应时间对形貌有一定的影响,5-240min都可以得到高质量的纳米晶,30-180min为最佳时间段。
具体地,有机钛酯的添加量对形貌有一定的影响,0.5-2ml都可以得到发光的CsPbBr3纳米晶,1ml为最佳添加量。
具体地,不同的有机钛酯的添加可以得到不同形貌的CsPbBr3纳米晶。钛酸四乙酯可以得到纳米片形貌的CsPbBr3纳米晶;钛酸异丙酯可以得到尺寸均一的纳米立方块形貌的CsPbBr3纳米晶;钛酸正四丁酯可以得到尺寸不均的纳米立方块形貌的CsPbBr3纳米晶。
本申请提供的CsPbBr3纳米晶的制备方法,其特征在于:反应过程只需一步,极其简便快捷。
本申请提供的CsPbBr3纳米晶的制备方法,其特征在于:有机钛酯起到了催化剂作用,并且改变其种类可以调节形貌。
本发明提供的CsPbBr3纳米晶的制备方法,其特征在于:可倍化合成,用于工业化生产。
根据本申请的又一方面,提供了一种发光材料,包含上述方法制备得到的钙钛矿纳米晶材料。
可选地,所述钙钛矿纳米晶材料的尺寸为10~100nm。
可选地,所述钙钛矿纳米晶材料的尺寸为15~25nm。
可选地,所述钙钛矿纳米晶材料的尺寸上限选自50nm、60nm、80nm、90nm或100nm;下限选自10nm、15nm、20nm、25nm或40nm。
可选地,所述钙钛矿纳米晶材料的发光波长在480~520nm。
可选地,所述钙钛矿纳米晶材料的发光波长上限选自490nm、495nm、500nm、509nm、510nm或520nm;下限选自480nm、485nm或490nm。
本申请中所有涉及数值范围的条件均可独立地选自所述数值范围内的任意点值。
本申请中“C1~C20”、“C1~C10”等均指基团所包含的碳原子数。
本申请中,“烷基”是由烷烃化合物分子上失去任意一个氢原子所形成的基团。
本申请中,“烃基”为烃分子中失去碳原子上的一个氢原子后形成的基团。所述烃为碳水化合物,例如烷烃、烯烃、炔烃均为烃。
本申请中,“芳基”为芳香族化合物分子中失去芳香环上任意一个氢原子后形成的基团。
本申请能产生的有益效果包括:
1)本申请所提供的钙钛矿微晶的制备方法,成功在室温条件下,在有机钛酯的催化作用下一步反应合成出了CsPbBr3纳米晶,为其工业化生产提供了可能,使其在光电器件领域具有广阔的应用前景。
2)本申请所提供的钙钛矿微晶的制备方法,具有可倍化合成,用于工业化生产。
3)本申请所提供的钙钛矿微晶的制备方法,反应过程只需一步,极其简便快捷,反应时间为30~180min。
4)本申请所提供的钙钛矿微晶的制备方法,有机钛酯起到了催化剂作用,并且改变其种类可以调节形貌。
附图说明
图1为实施例1中添加1ml的钛酸异丙酯,室温下搅拌60min得到的CsPbBr3纳米晶的XRD图。
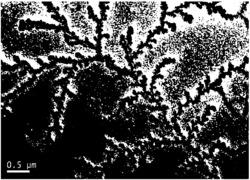
图2为实施例1中添加1ml的钛酸异丙酯,室温下搅拌60min得到的CsPbBr3纳米晶的透射电镜图,其中方框内表示竖立的纳米片。
图3为实施例2中添加1ml的钛酸四乙酯,室温下搅拌60min得到的CsPbBr3纳米晶的透射电镜图。
图4为实施例3中添加1ml的钛酸正四丁酯,室温下搅拌60min得到的CsPbBr3纳米晶的透射电镜图。
图5为实施例1-3中添加不同有机钛酯得到的样品的荧光光谱和光吸收谱图,其中(a)对应实施例1;(b)对应实施例2;(c)对应实施例3。
图6为实施例1、4-6中添加1ml的钛酸异丙酯,分别在室温下搅拌60、15、30、180min得到的CsPbBr3纳米晶的透射电镜图,其中(a)对应实施例4;(b)对应实施例5;(c)对应实施例1;(d)对应实施例6。
图7为实施例1、7-9中分别添加1、0.5、1.5、2ml的钛酸异丙酯,室温下搅拌60min得到的CsPbBr3纳米晶的透射电镜图,其中(a)对应实施例7;(b)对应实施例2;(c)对应实施例8;(d)对应实施例9。
具体实施方式
下面结合实施例详述本申请,但本申请并不局限于这些实施例。
如无特别说明,本申请的实施例中的原料和溶剂均通过商业途径购买。
本申请的实施例中分析方法如下:
利用Rigaku MiniFlex II X-射线粉末衍射仪进行XRD测试,测试范围:10-70度,扫描速率为2度/分,扫描步长为0.02度。
利用Tecnai G2S-Twin F20场发射透射电子显微镜进行形貌测试。
利用Varian Cary 500scan荧光光谱仪进行荧光性能测试。
利用日立UV-2450紫外可见光吸收谱仪进行紫外测试。
实施例1样品1
步骤1:将10ml十八烯加入到20ml的试剂瓶中,加入0.5ml的油酸和0.5ml的油胺,然后加入1ml的钛酸异丙酯,不断搅拌(以560每分钟的频率摇床振荡,摇床型号为 KS130basic);
步骤2:称量0.3mmol的PbBr2和0.1mmol的Cs2CO3加入到上述不断搅拌的溶液中,在室温及空气环境中,几分钟后即可观察到溶液颜色变为黄色,持续搅拌60min紫外灯(365nm)照射下可观察到明亮的绿色光;
步骤3:将反应后的溶液在11000rpm条件下离心5分钟,保留下层沉淀中的CsPbBr3纳米晶并用甲苯分散保存,得到的CsPbBr3纳米晶记为1
实施例2样品2
其他步骤同实施例1,不同之处在于步骤1中的钛酸异丙酯换为钛酸四乙酯,得到的CsPbBr3纳米晶记为2
实施例3样品3
其他步骤同实施例1,不同之处在于步骤1中的钛酸异丙酯换为钛酸正四丁酯,得到的CsPbBr3纳米晶记为3
实施例4样品4
其他步骤同实施例1,不同之处在于步骤2中的搅拌时间由60min改为15min,得到的CsPbBr3纳米晶记为4
实施例5样品5
其他步骤同实施例1,不同之处在于步骤2中的搅拌时间由60min改为30min,得到的CsPbBr3纳米晶记为5
实施例6样品6
其他步骤同实施例1,不同之处在于步骤2中的搅拌时间由60min改为180min,得到的CsPbBr3纳米晶记为6
实施例7样品7
其他步骤同实施例1,不同之处在于步骤1中的钛酸异丙酯的量由1ml变为0.5ml,得到的CsPbBr3纳米晶记为7
实施例8样品8
其他步骤同实施例1,不同之处在于步骤1中的钛酸异丙酯的量由1ml变为1.5ml,得到的CsPbBr3纳米晶记为8
实施例9样品9
其他步骤同实施例1,不同之处在于步骤1中的钛酸异丙酯的量由1ml变为2ml,得到的CsPbBr3纳米晶记为9
实施例10样品1
对样品1
实施例11样品1
对样品1
通过改变搅拌时间制备了形貌略有不同的CsPbBr3纳米晶,对CsPbBr3纳米晶进行了透射电镜测试。图6为四种不同搅拌时间下得到的透射电镜图片(对应实施例4~6),可以看出15min时的样品颗粒较小且厚度较薄,随着搅拌时间的增加,颗粒逐渐长成纳米立方块,伴随少量的纳米片。但从30min开始,颗粒的大小基本已经不变,保持在20nm左右。所以,增加搅拌时间不会使颗粒持续增长,30-180min都为合理的反应时间。
通过改变钛酸异丙酯的添加量调控了CsPbBr3纳米晶的形貌,对CsPbBr3纳米晶进行了透射电镜测试。图7为四种不同添加量下得到的透射电镜图片(对应实施例7~9),可以看出0.5ml时的样品颗粒尺寸很小且没有立方形貌,为无形貌的小颗粒。1ml的添加量时得到了很好的均一的立方块形貌。随着钛酸异丙酯的继续增加,颗粒表面覆盖了一些没有形貌的物质。这些物质是钛酸异丙酯水解产生的钛氧物质,从而使产物不是纯的CsPbBr3纳米晶。因此,最合适的添加量为1ml,此时能得到形貌好的纳米晶。
实施例12样品1
对样品1
根据以上结果,可以看出本发明提出的方法成功的快速制备出了形貌可调的CsPbBr3纳米晶,该CsPbBr3纳米晶合成的最佳时间为30~180min,最佳有机钛酯添加量为1ml。该方法填补了CsPbBr3纳米晶室温下合成技术的匮乏。本发明确定了CsPbBr3纳米晶的产业化生产基础条件。
实施例13实施例1~9的放大生产
对实施例1~9的反应物投放质量放大20倍后,仍然得到同样的效果。
本领域技术人员根据需要选择机械搅拌、磁力搅拌、高速分散、振荡的至少一种条件(速率为400~640rpm/min)均可实现本申请中的技术方案,达到所述技术效果。以上对本发明做了示例性的描述,应该说的是,在不脱离本发明的核心的情况下,任何简单的变形、修改或者其他本领域技术人员能够不花费创造性劳动的等同替换均落入本发明的保护范围。
本发明未尽事宜为公知技术。
以上所述,仅是本申请的几个实施例,并非对本申请做任何形式的限制,虽然本申请以较佳实施例揭示如上,然而并非用以限制本申请,任何熟悉本专业的技术人员,在不脱离本申请技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
一种钙钛矿纳米晶的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。




















动态评分
0.0