

IPC分类号 : C08G61/00,C08G61/02,C08G61/12,C08J3/28,C08J3/24,H01L51/50,H01L51/54





专利摘要
本发明提供含可交联基团的水醇溶共轭聚合物材料及其应用。所述共轭聚合物材料具有共轭的主链及功能化的侧链基团,其中功能化的侧链基团包括可交联的取代基团A及具有水醇溶性的强极性基团B。由于A、B功能化取代基的存在,所述共轭聚合物材料具有可用醇等强极性溶剂加工及在光照或者加热的条件下交联生成不溶不熔的互穿网络聚合物的特征,并且具有良好的增强高功函数金属电极电子注入的性能,适于制作复杂的多层有机电子器件。所述共轭聚合物能够作为电子注入/传输层应用在发光、光伏等有机光电器件中,改善器件性能。
说明书
技术领域
本发明涉及高分子光电材料领域,具体涉及含可交联基团的水醇溶共轭聚合物材料及其应用。
背景技术
自从1990年第一个聚合物电致发光二极管发明以来,聚合物电光电材料得到了学术界和工业界的广泛关注。
为了实现高效的聚合物电致发光器件,电子和空穴分别从阴极和阳极高效的注入是其中的关键。因此,很多高效的聚合物电致发光器件都是采用多层器件结构,即除了发光层外,还含有一层或多层空穴传输/注入层或电子传输/注入层。因此,除了开发优异的发光材料,开发优异的电子传输/注入材料和空穴传输/注入材料也是实现高效聚合物电致发光器件的关键。
我们此前的研究发现共轭聚电解质及其中性前驱体是一类非常优异的电子注入/传输材料(Chem. Mater. 2004, 16, 708.; Adv. Mater. 2004, 16, 1826;中国专利ZL200310117518.5)。这类材料在极性溶剂中有很好的溶解性,同时具有优异的电子传输性能,从而使得制备高效多层结构的聚合物电致发光器件成为可能。此外,这类材料还能有效的增加从高功函数的金属(如铝,银,金)向聚合物半导体的电子注入,更有利于以印刷的方式实现高分子多层器件(Adv. Mater. 2007, 19, 810.)。后续的研究表明,这类共轭聚电解质材料不但可用于发光器件,还可作为界面修饰层大幅提高有机太阳能电池、场效应晶体管的性能(Chem. Soc. Rev., 2010, 39, 2500)。
发明内容
本发明提供含交联基团的水醇溶共轭聚合物材料及其应用,这种材料不仅在光照或者加热的条件下相互反应生成不溶不熔的网状聚合物膜,而且还能通过其极性基团与金属阴极形成偶极子来降低电子的注入势垒和提高电子的传输能力。
本发明的含可交联基团的水醇溶共轭聚合物材料具有如下结构:
其中,n为1~10000的自然数,A为含有乙烯基的交联基团或含有环状烷氧基的交联基团,B为具有水醇溶性的强极性基团,C1和C2为相同或者不同的苯环衍生物或者含有碳碳双键、碳碳三键、碳氮键的共轭单元,R1、R2分别为A与C1、B与C2之间的连接单元,R1、R2为相同或者不同的C1~C20的烷基链,或者R1、R2为相同或者不同的C1~C20的烷基,其中烷基上一个或多个碳原子被氧原子、烯基、炔基、芳基或酯基中的一种以上官能团取代,氢原子被氟原子、氯原子、溴原子、碘原子或上述官能团取代。
上述强极性基团为胺基、季铵盐基团、磷酸根、磷酸酯基、磺酸根、羧基和羟基中的一种以上。
所述交联基团在在光照或者加热条件下能发生自由基反应、协同反应、亲电反应、亲核反应。
所述C1和C2分别具有如下结构的一种以上:
。
作为优选的,所述苯环衍生物为苯环、芴、咔唑或硅芴衍生物的一种以上,具有以下结构的一种以上:
。
所述含有环状烷氧基的交联基团其环状烷氧基为三元环、四元环烷氧基,具有以下结构的一种以上:
。
所述含有乙烯基的交联基团具有以下结构的一种以上:
。
所述烷基为直链烷基、支链烷基或者环状烷基。
上述环状烷氧基交联基团在光照、加热条件下能够发生交联,其交联机理主要是光酸在光照条件下产生酸氢离子,这种酸在加热的条件下使得环状烷氧基交联基团发生阳离子开环聚合物,如果R为聚合物时,就能形成具有抗溶剂性能的交联网状结构,以四元环烷氧基单元为例,其交联机理如下所示:
。
上述含乙烯基的交联基团在光照、加热条件下能够发生交联,其交联机理主要是交联基团在加热或者光照条件下,发生自由基聚合,或者协同反应(环化二聚),以苯基三氟乙烯基醚为例,其环化二聚机理如下所示:
。
以苯乙烯为例,其自由基聚合机理如下所示
。
在有机电致发光器件(ITO阳极/空穴传输层/发光层/电子传输层/铝阴极)中,上述含可交联基团取代的水醇溶共轭聚合物作为电子传输材料的应用。在发光共轭聚合物和阴极铝之间插入一层含有所述聚合物膜,在光照或加热条件下,将所述含可交联基团的水醇溶共轭聚合物材料用强极性溶剂加工成不溶不熔的互穿网络聚合物,作为金属电极电子注入层应用在有机电子器件中。该聚合物不仅具有抗溶剂性,而且能够降低电子的注入势垒,增强电子的注入效率,平衡双极载流子,使得空穴-电子在发光层中复合,达到改善辐射发光的效率。
与现有技术相比,本发明具有以下优点:
(1)本发明的聚合物在具有良好的水醇溶性的同时,还具有可交联的功能。在制备复杂多层器件时即可利用与传统油溶性光电高分子溶解性的不同,又可利用交联的方式进行多层器件加工,相比于传统的水醇溶或可交联光电材料具有更多的选择性;
(2)和传统的可交联光电材料相比,本发明聚合物的强极性基团使材料可用环境友好的溶剂(如水醇等)加工的同时,具有良好的电子注入性能(Chem. Soc. Rev., 2010, 39, 2500)。
附图说明
图1为实施例1所制备的经交联处理的PFN-C膜、未经交联以处理的PFN-C膜经四氢呋喃溶液洗脱处理后的吸光度曲线图;
图2为分别以实施例1、2、3所制备的聚合物PFN-C、PFN-S、PF-P-C作为电子传输层的基于绿光材料P-PPV的有机电致发光器件的电流效率-电流密度曲线图;
图3为分别以实施例1、2、3所制备的聚合物PFN-C、PFN-S、PF-P-C作为电子传输层的基于橙红光材料MEH-PPV的有机电致发光器件的电流效率-电流密度曲线图;
图4为分别以实施例1、2、3所制备的聚合物PFN-C、PFN-S、PF-P-C作为电子传输层的发绿光有机电致发光器件的归一化电致发光光谱图;
图5为分别以实施例1、2、3所制备的聚合物PFN-C、PFN-S、PF-P-C作为电子传输层的发橙红光有机电致发光器件的归一化电致发光光谱图。
具体实施方式
下面通过具体实施例对本发明作进一步的说明,其目的在于帮助更好的理解本发明的内容,具体包括材料合成、表征与器件制备,但这些具体实施方案不以任何方式限制本发明的保护范围。
所述含可交联基团的水醇溶共轭聚合物材料通用合成方法是先合成带有功能化基团的单体,通过过渡金属催化偶联的方法得到所述聚合物,通过控制反应时间、反应温度可以控制聚合物的分子量以及分散系数,合成路线如下所示:
。
带有功能化基团的单体合成主要采用以下两种路线:
路线一,合成路线如下所示,共轭单元的反应位点与卤代烃、醇、酚通过亲核、威廉姆森反应、加成反应、酯化反应将C1-C20烷基链、烷氧基链通过共价键与共轭单元相连,进而与功能化基团反应得到功能化的单体,实施例1的单体3采用此路线合成的:
。
路线二,合成路线如下所示,卤代烃、醇、酚和功能化基团单元通过亲核、威廉姆森反应、加成反应、酯化反应得到功能化的C1-C20烷基、烷氧基,这些功能化的烷基、烷氧基进一步与共轭单元的反应位点反应即可得到功能化的单体,实施例1的单体1采用此路线合成的:
。
实施例1
含可交联基团的水醇溶聚{2,7-[9,9’-二(3-乙基-3-(6-已基)甲基醚-氧杂环丁烷)芴]-共-2,7-[9,9’-二(6-N,N-二乙基胺基-己基)芴]}(简称为PFN-C)的制备
合成路线如下:
(1)单体1 [2,7-二溴-9,9’-二(3-乙基-3-(6-已基)甲基醚-氧杂环丁烷)芴]是按照文献[J Polym Sci A Polym Chem., 2007, 3, 388]公开的方法制备,单体3 [2,7-二(三亚甲基硼酸酯)-9,9’-二(6-N,N-二乙基胺基-己基)芴] 是按照“先进材料”[Adv. Mater., 2011, 23, 1665]公开的方法制备。
(2)聚{2,7-[9,9’-二(3-乙基-3-(6-已基)甲基醚-氧杂环丁烷)芴]-共-2,7-[9,9’-二(6-N,N-二乙基胺基-己基)芴]}的制备 (简称PFN-C)
将单体2,7-二(三亚甲基硼酸酯)-9,9’-二(6-N,N-二乙基胺基-己基)芴(728mg, 1mmol)、单体2,7-二溴-9,9’-二(3-乙基-3-(6-已基)甲基醚-氧杂环丁烷)芴(720mg, 1mmol)和10mg四合三苯基磷钯催化剂溶于10ml甲苯和5ml四氢呋喃的混合溶剂中,加入4ml 2mol/L的碳酸钠水溶液,在氩气的保护下,回流反应48小时,然后冷却到室温,将反应液在甲醇中沉淀得到粗品,将粗品溶于四氢呋喃中,过0.45μm的有机滤膜,浓缩,将浓缩后的溶液在甲醇中沉淀得到聚合物颗粒,用丙酮索氏提取器除去聚合物颗粒中的小分子物质后,最后将抽提得到的滤饼在真空烘箱中45℃下干燥24小时,得到832mg固体,即聚{2,7-[9,9’-二(3-乙基-3-(6-已基)甲基醚-氧杂环丁烷)芴]-共-2,7-[9,9’-二(6-N,N-二乙基胺基-己基)芴]}(简称PFN-C),其产率为70%。
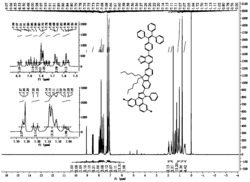
所得固体的核磁数据如下:1H NMR (300 MHz, CDCl3) δ (ppm) 7.95-7.75 (d, 4H), 7.75-7.60 (s, 8H), 4.50 and 4.33 (dd, 8H, four -CH2- in the oxetane ring), 3.45 (s, 4H), 3.32 (t, 4H), 2.51-2.44 (q, 8H), 2.30-2.28 (t, 4H), 2.04-1.80 (m, 8H), 1.70 (q, 4H), 1.41 (m, 4H), 1.30-0.90 (m, 28H), 0.84-0.53 (m, 8H)。
图1为实施例1所合成的聚合物PFN-C溶液(溶剂为二甲苯,另外加入1%的光酸)成膜后,在波长为365纳米的紫外光照射1分钟后,于160摄氏度加热板上加热15分钟使PFN-C膜交联,并且用四氢呋喃洗过后的UV吸光度曲线图。通过对图1分析可知,未交联时,用四氢呋喃溶剂洗过后,PFN-C膜的吸光度(曲线2)下降了40%;而紫外灯照射并且在加热板上加热,用四氢呋喃溶剂洗过后,PFN-C膜的吸光度(曲线2)几乎不下降,100%保持原有的吸光度。这说明PFN-C膜发生了交联,具有优良的抗溶剂性能。
实施例2
制备聚{2,7-[9,9’-二(3-乙基-3-(6-已基)甲基醚-氧杂环丁烷)芴]-共-2,7-[9,9’-二(6-N,N-二乙基胺基-己基)芴]}(简称为PFN-S)
合成路线如下:
(1)单体1 [2,7-二溴-9,9’-二(6-对醛基酚氧己基)芴]的制备
将原料2,7-二溴-9,9’-(6-溴己基)芴(32.5g, 50mmol)加入至反应瓶中,加入300ml N,N-二甲基甲酰胺(DMF)使原料溶解,然后再加入对羟基苯甲醛(15.3g, 125mmol)和碳酸钾2g,在氩气保护下加热回流12小时,冷却后将反应液倒入冰水中,经二氯甲烷萃取、浓缩后对浓缩物进行过柱,得到产物29.2g,产率为80%。
产物的核磁数据如下:1H NMR (300 MHz, CDCl3),δ (ppm): 9.86 (s, 2H), 7.82-7.78 (d, 4H), 7.77-7.51 (d, 2H), 7.50-7.44 (dd, 4H), 6.95-6.90 (d, 4H), 3.94-3.48 (t, 4H), 1.97-1.41 (m, 4H), 1.67-1.65 (m, 4H), 1.29-1.19 (m, 8H), 0.66-0.56 (m, 4H)。13C NMR (75MHz, CDCl3), δ (ppm): 190.72, 164.15, 152.23, 139.09, 131.93, 130.32, 129.79, 126.10, 121.55, 121.22, 114.71, 68.20, 55.09, 40.09, 29.48, 28.86, 25.58, 23.57。 Anal. calcd for C39H40Br2O2: C 63.94, H 5.50, Br 21.82, O, 8.74; found: C 63.85, H 5.36, Br 21.75, O, 9.04。
(2)单体2 [2,7-二溴-9,9’-二(6-乙烯基酚氧己基) 芴] 的制备
将witting试剂碘化甲基三苯基磷(7g, 25mmol)和叔丁醇钠(2.4g, 30mmol)加入至反应瓶中,加入100ml干燥的四氢呋喃,冰浴下反应30分钟。之后往反应液中加入2,7-二溴-9,9’-二(6-对醛基酚氧己基)芴(7.3g, 10mmol),让其在室温下反应2小时。将反应液倒入冰水中,二氯甲烷萃取、浓缩后进行过柱,经甲醇重结晶后得到产物6.17g,产率为85%。
产物的核磁数据如下:1H NMR (300 MHz, CDCl3), δ (ppm): 7.53-7.52 (d, 2H), 7.50-7.49 (d, 4H), 7.46-7.29 (d, 4H), 7.25-6.77 (d, 4H), 6.68-6.59 (m, 2H), 6.68-6.59 (d, 2H), 5.61-5.55 (d, 2H), 5.11-5.08 (d, 2H), 3.85-3.81 (t, 4H), 1.95-1.90 (m, 4H), 1.61-1.54 (m, 4H), 1.25-1.12 (m, 8H), 0.63-0.59 (m, 4H)。13C NMR (75MHz, CDCl3),δ (ppm): 158.87, 152.32, 139.08, 136.28, 130.28, 130.24, 127.32, 126.13, 121.54, 121.20, 114.47, 111.38, 67.83, 55.61, 40.10, 29.69, 29.09, 25.66, 23.61。 Anal. calcd for C41H44Br2O2: C 67.59, H 6.09, Br 21.93, O, 4.39; found: C 67.85, H 5.46, Br 21.75, O, 4.94。
(3)聚{2,7-[9,9’-二(3-乙基-3-(6-已基)甲基醚-氧杂环丁烷)芴]-共-2,7-[9,9’-二(6-乙烯基酚氧己基)]芴}(简称为PFN-S)
将实施例1中制备的单体2,7-二(三亚甲基硼酸酯)-9,9’-二(6-N,N-二乙基胺基-己基)芴(728mg, 1mmol)、单体2,7-二溴-9,9’-二(6-乙烯基酚氧己基)芴(728mg, 1mmol)和10mg四合三苯基磷钯催化剂溶于10ml 甲苯和5ml 四氢呋喃的混合溶剂中,加入4ml 2mol/L的碳酸钠水溶液,在氩气的保护下,回流反应48小时,然后冷却到室温,将冷却后的反应液在甲醇中沉淀得到粗品,将粗品溶于四氢呋喃中过100目的硅胶柱,之后过0.45μm的有机滤膜,浓缩,将浓缩后的溶液在甲醇中沉淀得到聚合物颗粒,用丙酮索氏提取器除去聚合物颗粒中的小分子物质后,将所得的滤饼于真空烘箱中45℃下干燥24小时,得到832mg固体,产率为70%。
所得固体的核磁数据如下:1H NMR (300 MHz, CDCl3) δ (ppm) 7.55-7.52 (d, 2H), 7.50-7.49 (d, 4H), 7.48-7.47 (d, 4H), 7.46-7.29 (d, 4H), 7.25-6.77 (d, 4H), 6.68-6.59 (m, 2H), 6.68-6.59 (d, 2H), 5.61-5.55 (d, 2H), 5.11-5.08 (d, 2H), 3.85-3.81 (t, 4H), 2.51-2.44 (q, 8H), 2.30-2.28 (t, 4H), 1.95-1.90 (m, 8H), 1.61-1.54 (m, 4H), 1.30-1.26 (m, 4H), 1.25-1.12 (m, 8H), 1.09-1.08 (m, 8H), 1.07-0.96 (t, 12H), 0.63-0.59 (m, 8H)。
实施例3
制备聚{2,7-[9,9’-二(3-乙基-3-(6-已基)甲基醚-氧杂环丁烷)芴]-共-2,7-[9,9’-二(6-磷酸二乙酯-己基)芴]}(简称为PF-P-C)
合成路线如下:
(1)2,7-二溴[9,9’-二(6-磷酸二乙酯-己基)芴]
该化合物是根据“大分子”(Macromolecules, 2005, 13, 5416)公开的方法制备。
(2)2,7-二(三亚甲基硼酸酯)[9,9’-二(3-乙基-3-(6-已基)甲基醚-氧杂环丁烷)]芴
将2,7-二溴-9,9’-二(3-乙基-3-(6-已基)甲基醚-氧杂环丁烷)芴(1.32g, 1.9mmol)加入至反应瓶中,加入20ml四氢呋喃溶解,在-78℃氩气氛围保护下加入3.5ml 2.5mol/L的正丁基锂正己烷溶液,-78℃保温反应2小时,加入1.44g氧杂硼烷(7.6mmol),反应24小时,让其自然升至室温,将反应液倒入冰水中,二氯甲烷萃取,无水硫酸镁干燥、浓缩后进行过柱分离,再经正己烷重结晶得到1.2g白色固体,产率为80%。
所得产物的核磁数据为:1H NMR (300 MHz, CDCl3), δ (ppm): 7.82-7.79 (2H, d, J = 7.5 Hz), 7.7 (2H, s), 7.71- 7.69 (2H, d, J = 7.5 Hz), 4.44-4.42 (4H, d, J =5.7 Hz), 4.30-4.28 (4H, d, J=5.7 Hz), 3.37 (4H, s), 3.32-3.28 (4H, t, J = 6.6 Hz), 1.95 (4H, m), 1.39-1.24 (10H), 1.04 (8H, m), 0.61-0.55 (4H, m)。13C NMR (75 MHz, CDCl3): 150.26, 143.87, 133.69, 128.77, 119.40, 83.73, 80.24, 75.98, 71.53, 55.08, 40.14, 39.79, 29.81, 29.42, 25.73, 24.92, 21.33。 Anal. Calcd for C47H72B2O8: C, 71.76, H, 9.22; Found: C, 71.27, H, 9.02。
(3)聚{2,7-[9,9’-二(3-乙基-3-( 6-已基)甲基醚-氧杂环丁烷)芴]-共-2,7-[9,9’-二(6-磷酸二乙酯-己基)芴]}(简称为PF-P-C)
将单体2,7-二(亚硼酸酯)-9,9’-二(3-乙基-3-(6-已基)甲基醚-氧杂环丁烷)芴(806mg,1mmol)、2,7-二溴[9,9’-二(6-磷酸二乙酯-己基)芴] (764mg,1mmol)和10mg四合三苯基磷钯催化剂溶于10ml 甲苯和5ml 四氢呋喃的混合溶剂中,加入4ml 2mol/L的碳酸钠水溶液,在氩气的保护下,回流反应48小时,然后冷却到室温,将反应液在石油醚中沉淀得到粗品,将粗品溶于四氢呋喃中过60-100目的硅胶柱,之后过0.45μm的有机滤膜,浓缩,将此溶液在石油醚中沉淀得到聚合物颗粒,用丙酮索氏提取器除去小分子物质,最后在真空烘箱中45℃下干燥24小时,得到689mg固体,产率为60%。
所得固体的核磁数据如下:1H NMR (300 MHz, CDCl3), δ (ppm): 7.83-7.70 (4H, m,), 7.68-7.47 (8H, m), 4.44-4.38 (8H, dd), 4.02-4.00 (8H, d), 3.37 (4H, s); 3.32-3.28 (4H, t), 2.11-1.88 (8H, m), 1.69-1.65 (8H, m), 1.56-1.48 (8H, m), 1.27-1.17 (28H, m), 0.85-0.80 (14H, m)。
实施例4
制备聚{2,7-[9,9’-二(3-乙基-3-(6-已基)甲基醚-氧杂环丁烷)芴]-共-4,4’-二苯基-1,3,4-恶二唑}(简称为PF-OXD-C)
合成路线如下:
将单体2,7-二(亚硼酸酯)-9,9’-二(3-乙基-3-(6-已基)甲基醚-氧杂环丁烷)芴(806mg, 1mmol)、2,5-二(4-溴苯基)-1,3,4-恶二唑(380mg, 1mmol)和10mg四合三苯基磷钯催化剂,溶于10ml 甲苯和5ml 四氢呋喃的混合溶剂中,加入4ml 2mol/L的碳酸钠水溶液,在氩气的保护下,回流反应48小时,然后冷却到室温,将反应液在甲醇中沉淀得到粗品,将粗品溶于四氢呋喃中过60-100目的硅胶柱,之后过0.45μm的有机滤膜,浓缩,将此溶液在甲醇中沉淀得到聚合物颗粒,用丙酮索氏提取器除去小分子物质,最后在真空烘箱中45℃下干燥24小时,得到832mg固体,产率为70%。
所得固体的核磁数据如下:1H NMR (300 MHz, CDCl3) δ (ppm) 7.98-7.97 (d, 4H), 7.70-7.66 (d, 4H) 7.52 (d, 2H), 7.50 (d 2H), 7.42 (d ,2H), 4.33 和4.50 (dd, 4H, two -CH2 –in the oxetane ring), 3.45 ( s ,4H), 3.32 (t ,5H), 2.04 (d ,4H), 1.91 (m ,4H),1.70 (m ,4H), 1.37 (m, 2H), 1.25 (t, 2H), 1.09(m ,4H), 0.84 (m, 6H), 0.57 (m ,4H)。
实施例5
以实施例1所合成的聚合物PFN-C为例说明此类聚合物交联处理后具有抗溶剂洗脱的性能
将PFN-C在对二甲苯中溶解,加入质量分数为1%的光酸[2-(4-甲氧基苯乙烯基)-4,6-双(三氯甲基)-1,3,5-三嗪],其中光酸的作用是在紫外光照射下提供酸,使氧杂环氧丁烷发生阳离子开环聚合。以0.45μm有机滤膜过滤,在普通玻璃片上旋涂成膜,厚度大约为20纳米。用惠普公司生产的UV测试仪(HP 8453 spectrophotometer)测PFN-C成膜后的吸光度,对应于图1中的曲线1。之后将PFN-C膜在波长为365纳米的紫外光下照射1分钟,于加热板上加热(160℃)15分钟使交联基团氧杂环氧丁烷发生开环聚合形成不溶不熔的交联网状膜。再用THF洗脱刚交联过的PFN-C膜,通过UV测试洗脱后的PFN-C膜的吸光度,对应于图1中的曲线3。同时对比了,未交联的PFN-C的抗溶剂洗脱性能,对应于图1中的曲线2。通过观察用THF洗脱后PFN-C膜吸光度的下降程度,就可看出交联程度,吸光度下降越多,表明大部分聚合物被THF洗掉,交联度低;吸光度下降越少,则说明大部分聚合物不能被THF洗掉,交联度高。通过对图1分析可知,未交联时,用THF溶剂洗过后,PFN-C膜的吸光度(曲线2)下降了40%,而光照、加热交联后,用THF溶剂洗脱后,PFN-C膜的吸光度几乎不下降(曲线3),100%保持原有的吸光度。这说明PFN-C膜发生了交联,具有优良的抗溶剂洗脱性能。
实施例6
实施例1、例2、例3所制备的聚合物PFN-C、PFN-S、PF-P-C作为电子传输材料在有机电致发光器件(ITO阳极/空穴传输层/发光层/电子传输层/铝阴极)中的应用
将ITO导电玻璃、约20欧/平方厘米得方块电阻预切成15毫米×15毫米方片。依次用丙酮、微米级半导体专用洗涤剂、去离子水、异丙醇超声清洗,氮气吹哨后置于恒温烘箱备用。使用前,ITO玻璃片在氧等离子体刻蚀仪中以等离子体轰击10分钟。PEDOT:PSS水分散液(约1%)购自Bayer公司,缓冲层以匀胶机(KW-4A)高速旋涂,厚度由溶液浓度和转速决定,用表面轮廓仪(Tritek公司Alpha-Tencor-500型)实测监控。成膜后,于恒温真空烘箱中驱除溶剂残余、竖膜。在ITO基片上甩PEDOT:PSS的膜厚40纳米左右为佳。
将荧光共轭聚合物P-PPV(P-PPV为发绿光材料)或MEH-PPV(MEH-PPV为发橙红光材料)于干净瓶中称量后,转入氮气保护成膜专用手套箱(VAC公司),在甲苯中溶解,以0.45微米滤膜过滤。在PEDOT:PSS膜上甩荧光聚合物,聚合物发光层最佳厚度为70~90纳米。膜厚用Alpha-Tencor-500表面轮廓仪测定。以1%的PFN-C、PFN-S或PF-P-C的甲醇:乙酸(100:1)溶液,在聚合物发光层上旋涂电子传输材料。对于PFN-C、PF-P-C材料需往溶液中加入1%的光酸,并且成膜后在波长为365纳米的紫外灯照射1分钟(仅对PFN-C、PF-P-C有效)。于120℃加热板上对器件进行热处理15分钟。在电子传输层上真空蒸镀铝(100纳米)作阴极。器件的发光区域有掩膜于ITO交互盖的区域确定为0.15平方厘米。所有制备过程均在提供氮气氛围的手套箱内进行。器件的电流-电压特性,发光强度和外量子效率由Keithley236电流电压-测量系统及一个经校正的硅光二极管测得。为显示本发明所采用的电子传输层的效果,在发光层上真空蒸镀Ba/Al作为阴极,或者在发光层上真空蒸镀Al作为阴极。测量结果如表1所示,PFN-C、PFN-S、PF-P-C作为电子传输层器件的电致发光器件结构ITO /PEDOT 4083 / Active Layer (MEH-PPV or P-PPV, 80nm)/ ETL / Al (~100nm)。其中I为 P-PPV,II为 MEH-PPV,LEmax:最大电流效率,LE:电流效率,V电压,Von:亮度为1坎德拉/平方米时的电压,Lmax:最大亮度,J:电流密度。
表1
由此可见,对于P-PPV发绿光器件、或者MEH-PPV发橙红光器件来说,在发光层与阴极之间插入一层本发明所述的可交联的、醇溶性的电子传输材料PFN-C、PFN-S、PF-P-C之后,器件的性能远高于只有Al阴极的器件,其器件性能可与结构为ITO/PEDOT:PSS/P-PPV or MEH-PPV/Ba/Al的器件相比,但是该器件使用了低功函数的金属Ba,容易被空气中的氧气氧化。尤其是PFN-C作为电子传输材料,绿光器件的最大电流效率达到12.6坎德拉/安培,相对只有Al阴极的器件来说,电流效率提高近60倍。在橙红光器件中,加入PFN-C之后,电流效率(0.73坎德拉/安培)增加更显著,比Ba/Al作阴极器件的电流效率(0.70坎德拉/安培)还高。
图2为以实施例1、2、3所合成的聚合物PFN-C、PFN-S、PF-P-C为电子传输层的发绿光的有机电致发光器件的电流效率-电流密度曲线图,器件结构为ITO/PEDOT:PSS/P-PPV /PFN-C or PFN-S or PF-P-C/Al。该器件电流效率与对比器件(ITO/PEDOT:PSS/P-PPV/Al)相比,升高很显著,这说明本发明所述的聚合物具有很好的电子注入能力和电子传输效率。其中以PFN-C为电子传输层的器件最大电流效率提高接近60倍;与对比器件(ITO/PEDOT:PSS/P-PPV/Ba/Al)相比,最大电流效率很接近,随着电流密度的增加,电流效率下降不明显。但是Ba阴极容易被氧化,极不稳定。
图3为以实施例1、2、3所合成的聚合物PFN-C、PFN-S、PF-P-C为电子传输层的发橙红的有机电致发光器件的电流效率-电流密度曲线图,器件结构为ITO/PEDOT:PSS/MEH-PPV /PFN-C or PFN-S or PF-P-C/Al。该器件电流效率与对比器件(ITO/PEDOT:PSS/MEH-PPV/Al)相比,升高也很显著,同样说明了本发明所述的聚合物具有很好的电子注入能力和电子传输效率。对于PFN-C而言,其作用与活泼金属Ba相当,而PFN-C很稳定。
图4为以实施例1、2、3所合成的聚合物PFN-C、PFN-S、PF-P-C为电子传输层的发绿光的传统有机电致发光器件的归一化电致发光光谱,器件结构为ITO/PEDOT:PSS/P-PPV /PFN-C or PFN-S or PF-P-C/Al。使用不同的阴极材料,对绿光器件而言,发射光谱基本相同,说明电子和空穴很好地限制在发光层P-PPV中。
图5为以实施例1、2、3所合成的聚合物PFN-C、PFN-S、PF-P-C为电子传输层的发橙红光的传统有机电致发光器件的归一化电致发光光谱,器件结构为ITO/PEDOT:PSS/MEH-PPV /PFN-C or PFN-S or PF-P-C/Al。使用不同的阴极材料,对橙红器件而言,发射光谱基本相同,说明电子和空穴很好地限制在发光层MEH-PPV中。
含可交联基团的水醇溶共轭聚合物材料及其应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。


















动态评分
0.0