










专利摘要
本发明涉及有机合成和药物化学领域,具体涉及一类结构新颖的夫西地酸衍生物及其制备方法和应用,优选的衍生物为:3β‑[(2‑氨基)乙酰氧基]‑21‑夫西地酸苄酯、3β‑[(2‑氨基)丙酰氧基]‑21‑夫西地酸苄酯、3β‑[(2‑氨基‑4‑甲基)戊酰氧基]‑21‑夫西地酸苄酯、3β‑(3‑氨基丙酰氧基)‑21‑夫西地酸苄酯、3β‑(4‑氨基丁酰氧基)‑21‑夫西地酸苄酯、3β‑(8‑氨基辛酰氧基)‑21‑夫西地酸苄酯、3β‑(11‑氨基十一碳酰氧基)‑21‑夫西地酸苄酯、3β‑(L‑赖氨酰氧基)‑21‑夫西地酸苄酯。本发明的夫西地酸衍生物具有抗肿瘤作用且作用机制明确,可以在开发新型抗肿瘤药物中得到应用。
权利要求
1.夫西地酸衍生物及其医学上可接受的盐,其特征在于所述夫西地酸衍生物为:
3α-[(2-氨基)乙酰氧基]-16β-乙酰氧基-11α-羟基-4α,8α,14β-三甲基-18-去甲-5α,10β-胆甾-(17Z)-17(20),24-二烯-21-酸苄酯;
3α-[(2-氨基)丙酰氧基]-16β-丙酰氧基-11α-羟基-4α,8α,14β-三甲基-18-去甲-5α,10β-胆甾-(17Z)-17(20),24-二烯-21-酸苄酯;
3α-[(2-氨基-4-甲基)戊酰氧基]-16β-戊酰氧基-11α-羟基-4α,8α,14β-三甲基-18-去甲-5α,10β-胆甾-(17Z)-17(20),24-二烯-21-酸苄酯;
3α-(3-氨基丙酰氧基)-16β-乙酰氧基-11α-羟基-4α,8α,14β三甲基-18-去甲-5α, 10β胆甾-(17Z)-17(20),24-二烯-21-酸苄酯;
3α-(11-氨基十一碳酰氧基)-16β-乙酰氧基-11α-羟基-4α,8α,14β三甲基-18-去甲-5α,10β胆甾-(17Z)-17(20),24-二烯-21-酸苄酯;
3α-[(2S)-2,6-二氨基己酰氧基]-16β-乙酰氧基-11α-羟基-4α,8α,14β三甲基-18-去甲-5α,10β胆甾-(17Z)-17(20),24-二烯-21-酸苄酯。
说明书
技术领域
本发明涉及有机合成和药物化学领域,具体涉及一类结构新颖的夫西地酸衍生物,含有它们的药物组合物及其制备方法。
技术背景
癌症是一种特别复杂,广泛和致命的疾病。2012年全球共新增1410万癌症案例并有820万人死亡,癌症死亡人数预计将持续增加,估计到2030年将有1310万人死亡。癌症已经成为严重危害人类健康和生命的重大疾病,迄今仍没有非常满意的治疗方法。而化学治疗已成为癌症治疗最重要的治疗方法之一。开发结构新颖的治疗药物至关重要。
夫西地酸(Fusidic acid,FA)是一种基于类固醇的具有四环体系的窄谱抑菌抗生素,它于1960年首次从真菌Fusidium coccineum中分离出来,自1962年开始用于由金黄色葡萄球菌以及其他几种革兰氏阳性菌引起的皮肤感染,骨关节感染和烧伤感染等的治疗。此外,夫西地酸是一种上市的抗菌药物,因此已被人们广泛研究并且已知其不具有细胞毒性。尽管研究者对夫西地酸进行了广泛研究,但未曾有夫西地酸衍生物可以用于临床。
公开的应用,夫西地酸临床上仅作为抗菌药物使用,无其他适应症,文献报道的夫西地酸衍生物也无抗肿瘤活性。
发明内容
本发明的目的就是提供一种结构新颖的夫西地酸衍生物及其制备方法,同时提供该类衍生物在抗肿瘤领域中的新应用,对开发新型抗肿瘤药物具有重要意义。
本发明是通过以下技术方案来实现:
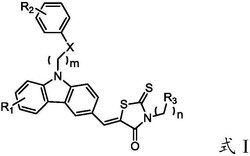
通式I、通式II所示夫西地酸衍生物及其医学上可接受的盐:
其中,
通式I:R代表H、甲基、异丙基、异丁基、苄基、(CH2)mNH2,其中,m=1-10,并且m为自然数;
通式II:n=1-10,并且n为自然数。
优选,所述化合物及其医学上可接受的盐,其中,
通式I:R代表H、甲基、异丙基、异丁基、苄基、(CH2)4NH2;
通式II:n=1、2、4、6、9。
优选,本发明的部分化合物为:
3β-[(2-氨基)乙酰氧基]-21-夫西地酸苄酯;
3β-[(2-氨基)丙酰氧基]-21-夫西地酸苄酯;
3β-[(2-氨基-4-甲基)戊酰氧基]-21-夫西地酸苄酯;
3β-(3-氨基丙酰氧基)-21-夫西地酸苄酯;
3β-(4-氨基丁酰氧基)-21-夫西地酸苄酯;
3β-(8-氨基辛酰氧基)-21-夫西地酸苄酯;
3β-(11-氨基十一碳酰氧基)-21-夫西地酸苄酯;
3β-(L-赖氨酰氧基)-21-夫西地酸苄酯;
药理试验与作用机制研究证明,本发明的夫西地酸衍生物具有抗肿瘤作用且作用机制明确,可以在开发新型抗肿瘤药物中得到应用。
所述的抗肿瘤作用为抗宫颈癌Hela肿瘤、抗多药耐药口腔表皮样癌KBV肿瘤、抗神经胶质瘤U87肿瘤、抗胃癌MKN45肿瘤、抗肝癌JHH-7肿瘤。
所述夫西地酸衍生物及其上述化合物的光学异构体或其药学上可接受的溶剂合物。
所述衍生物的制备方法如下。
通式I的化合物按如下方法合成制备:
a.以夫西地酸为原料,在无机碱存在下,溴化苄保护21-COOH;
b.在碱性条件下,Boc酸酐保护长链末端带氨基的酸;
c.在有机碱,缩合剂存在下,将步骤a所得的产物与Boc-氨基酸反应或将a、b两步所得的产物进行反应;
d.在酸性条件下脱除Boc。
通式II的化合物按如下方法合成制备:
a.以夫西地酸为原料,在无机碱存在下,溴化苄保护21-COOH;
b.在碱性条件下,Boc酸酐保护长链末端带氨基的酸;
c.在有机碱,缩合剂存在下,将a、b两步所得的产物进行反应;
d.在酸性条件下脱除Boc。
有益效果:本专利中具有抗肿瘤活性的夫西地酸衍生物属于首创,所述夫西地酸衍生物用于制备抗肿瘤药物。
夫西地酸是一种上市的抗菌药物,其本身无抗肿瘤活性,并且未有报道发现其衍生物具有抗肿瘤活性。而本发明提供的一类夫西地酸衍生物具有明显的抗肿瘤活性。
在5μM浓度下,本发明提供的一类夫西地酸衍生物对多种肿瘤细胞具有抑制作用,其中,实施例4对Hela、KBV、U87、MKN45四种肿瘤细胞都具有较强的抑制作用,存活率分别为13%、12%、9%、5%;在1.5μM浓度下,实施例4仍对四种肿瘤细胞具有较强的抑制作用,存活率分别为32%、14%、10%、23%,进一步的药理验证发现实施例4对Hela、U87、JHH-7、KBV、MKN45五种肿瘤细胞具有较强的抗肿瘤活性,IC50=1.26-3.57μM。
本发明化合物专利性在于提出了一类结构新颖的具有抗肿瘤活性且作用机制明确的化合物,进一步分析作用机制在于夫西地酸衍生物在1-10μM范围内以剂量依赖性的方式降低Hela细胞中新合成蛋白质的含量,导致细胞阻滞在细胞周期的Sub-G0/G1期,从而诱导Hela细胞凋亡。
附图说明
图1为实施例4对Hela细胞凋亡早期的影响。
图2为实施例4对Hela细胞凋亡晚期的影响。
图3为实施例4对Hela细胞蛋白质合成的影响。
具体实施方式
1.下面通过实施例进一步详细描述本发明,但本发明不仅仅局限于以下实施例。
实施例1
3β-[(2-氨基)乙酰氧基]-21-夫西地酸苄酯
取500mL茄型瓶,将夫西地酸(10.01g,0.019mol)溶于丙酮(200mL),搅拌加入碳酸钾(5.36g,0.039mol)、溴化苄(2.78mL,0.023mol),30℃反应5-7小时。抽滤,浓缩,乙酸乙酯(50mL)稀释,10%盐酸洗至酸性,水洗,饱和食盐水洗,无水硫酸钠干燥,过滤,减压蒸除溶剂,硅胶柱层析(V氯仿:V甲醇=210:1-190:1),得白色固体21-夫西地酸苄酯(8.86g,75.4%)。
取25mL的茄形瓶,将21-夫西地酸苄酯(0.10mmol)溶于无水二氯甲烷(10mL),搅拌加入Boc保护的氨基酸(0.22mmol),DMAP(0.31mmol),EDCI(0.30mmol),室温反应4-6小时。10%盐酸洗至酸性,水洗,饱和食盐水洗,无水硫酸钠干燥,过滤,减压蒸除溶剂,硅胶柱层析(V氯仿:V甲醇=400:1-200:1),得目标中间体X1-X3。
取25mL的茄形瓶,将X1(45mg,0.059mmol)溶于乙酸乙酯(10mL),逐滴加入37%盐酸(20:1),35℃搅拌2-6小时。依次用饱和碳酸氢钠溶液、水、饱和食盐水各洗涤三次,无水硫酸钠干燥,过滤,减压蒸除溶剂。硅胶柱层析(V氯仿:V甲醇=100:1-60:1),得白色固体(31mg,80.3%)。
实施例2
3β-[(2-氨基)丙酰氧基]-21-夫西地酸苄酯
以X2为原料,参照3β-[(2-氨基)乙酰氧基]-21-夫西地酸苄酯的制备方法,得白色固体(46mg,86.7%)。
实施例3
3β-[(2-氨基-4-甲基)戊酰氧基]-21-夫西地酸苄酯
以X3为原料,参照3β-[(2-氨基)乙酰氧基]-21-夫西地酸苄酯的制备方法,得白色固体(28mg,79.4%)。
实施例4
3β-(3-氨基丙酰氧基)-21-夫西地酸苄酯
取25mL的茄形瓶,将氢氧化钠(170mg,4.25mmol)水溶液(1.96mL)和Boc酸酐(915mg,4.19mmol)加入到叔丁醇中,然后加入相应的末端带氨基的酸(3.81mmol),室温搅拌18-24小时。加入水和1mol/L盐酸稀释,乙酸乙酯快速萃取,有机相水洗,饱和食盐水洗,无水硫酸钠干燥,抽滤,减压蒸除溶剂,得Boc保护的化合物X4-X8。
取25mL的茄形瓶,将X4-X8(0.10mmol)溶于二氯甲烷(8mL),加入DMAP(0.20mmol)和EDCI(0.20mmol),室温搅拌1小时后加入21-夫西地酸苄酯,继续反应20-24小时。减压除溶剂,乙酸乙酯(10mL)稀释,水洗,饱和食盐水洗,无水硫酸钠干燥,过滤,减压蒸除溶剂,柱层析(V石油醚:V乙酸乙酯=20:1-8:1),得目标中间体X9-X13。
取25mL的茄形瓶,将X9(35mg,0.045mmol)溶于无水二氯甲烷(10mL),冰浴下加入三氟乙酸(0.86mL),室温反应3-5小时。减压蒸除溶剂,柱层析(V二氯甲烷:V甲醇=100:1-50:1),得黄色固体(26mg,85.0%)。
实施例5
3β-(4-氨基丁酰氧基)-21-夫西地酸苄酯
以X10与三氟乙酸为原料,参照3β-(3-氨基丙酰氧基)-21-夫西地酸苄酯的制备方法,得到白色固体(29mg,73.6%)。
实施例6
3β-(8-氨基辛酰氧基)-21-夫西地酸苄酯
以X11与三氟乙酸为原料,参照3β-(3-氨基丙酰氧基)-21-夫西地酸苄酯的制备方法,得到白色固体(25mg,81.1%)。
实施例7
3β-(11-氨基十一碳酰氧基)-21-夫西地酸苄酯
以X12与三氟乙酸为原料,参照3β-(3-氨基丙酰氧基)-21-夫西地酸苄酯的制备方法,得到白色固体(27mg,78.8%)。
实施例8
3β-(L-赖氨酰氧基)-21-夫西地酸苄酯
以X13与三氟乙酸为原料,参照3β-(3-氨基丙酰氧基)-21-夫西地酸苄酯的制备方法,得到白色固体(28mg,80.5%)。
2.下面是本发明部分化合物的体外抑制肿瘤细胞活性实验。
(1)实验方法:
细胞培养与处理:
肿瘤细胞系:宫颈癌Hela细胞系、多药耐药口腔表皮样癌KBV细胞系、胶质瘤U87细胞系、胃癌MKN45细胞系、肝癌JHH-7细胞系。将所有细胞在37℃、5%CO2条件下的含有10%的热灭活胎牛血清、100U/mL青霉素和100g/mL链霉素的DMEM培养基中培养。指数生长期的细胞将用于进一步的实验。
MTT法:
将肿瘤细胞接种到96孔板中,总体积为100μL,密度为1×10
(2)实验结果:
表1.夫西地酸衍生物的体外肿瘤抑制活性。
由此表可知,在5μM浓度下,实施例1-实施例8对多种肿瘤细胞具有抑制作用,其中实施例4-实施例8对Hela、KBV、U87、MKN45四种肿瘤细胞都具有较强的抑制作用,存活率≤23%;在1.5μM浓度下,实施例4仍对四种肿瘤细胞具有较强的抑制作用,存活率分别为32%、14%、10%、23%。因此本发明进一步对实施例4进行多种肿瘤细胞体外抗肿瘤活性测定(见表2)。
表2.实施例4抑制多种肿瘤细胞的IC50值
由此表可知,夫西地酸无抗肿瘤活性,而实施例4对Hela、U87、JHH-7、KBV、MKN45五种肿瘤细胞具有较强的抗肿瘤活性,IC50=1.26-3.57μM。
3.实施例4的作用机制研究
(1)实验方法:
TUNEL染色:
将Hela细胞(2×10
细胞周期分析:
将Hela细胞(1×10
蛋白质合成实验:
将Hela细胞用10μg/mL的嘌呤毒素处理10分钟,用冰冷的磷酸缓冲盐溶液洗涤两次后,用SDS-PAGE样品缓冲液裂解细胞,在SDS-PAGE中分离出蛋白质并转移到Immobilon-P膜上。将膜在含有5%脱脂乳和0.1%吐温-20的Tris缓冲盐溶液中封闭处理1小时,然后加入嘌呤毒素抗体(1:1000稀释),在4℃下温育过夜。然后加入HRP缀合的抗小鼠IgG(1:5000稀释),在室温下温育1小时后,使用增强的化学发光使膜可视化。
(2)实验结果:
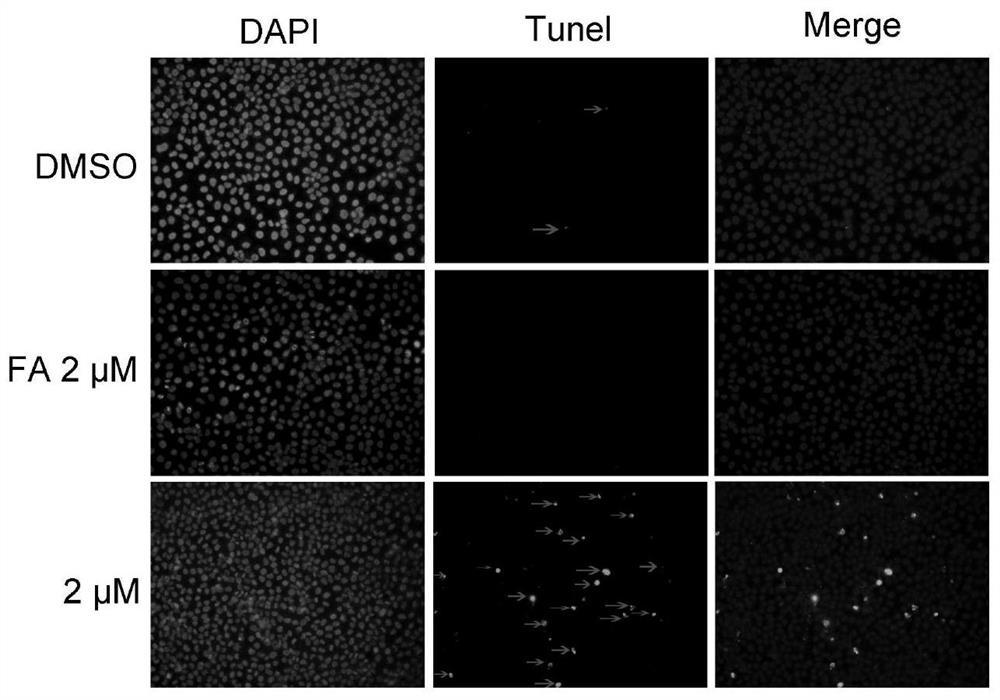
结果表明2μM的实施例4可以诱导Hela细胞凋亡,如附图1所示Hela细胞凋亡早期的影响,箭头表示TUNEL阳性细胞。
如附图2所示对Hela细胞凋亡晚期的影响,结果表明在48小时或72小时温育后,5μM的实施例4可以显着增加Sub-G0/G1期细胞的比例。
附图3为实施例4对Hela细胞蛋白质合成的影响。由于嘌呤霉素可以掺入新合成的蛋白质中,抗嘌呤霉素抗体可用于检测实施例4对新合成蛋白质的影响。附图3(A)结果显示5μM的实施例4降低了掺入嘌呤霉素的新合成蛋白质的含量,这表明实施例4可以抑制蛋白质的合成。附图3(B)表明实施例4可在1-10μM范围内以剂量依赖性的方式降低Hela细胞中新合成蛋白质的含量。
作用机制研究结果表明实施例4可在1-10μM范围内以剂量依赖性的方式降低Hela细胞中新合成蛋白质的含量,导致细胞阻滞在细胞周期的Sub-G0/G1期,从而诱导Hela细胞凋亡。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种等同变换,这些等同变换均属于本发明的保护范围。另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合。为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
具有抗肿瘤活性的夫西地酸衍生物及其合成制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。

















动态评分
0.0