

IPC分类号 : C07D313/00,A61K31/365,C07F7/18,A61P35/00





专利摘要
本发明涉及大环内酯Callyspongiolide立体异构体的合成及抗癌应用,利用经典的逆合成分析,成功合成了Callyspongiolide天然产物以及它的三个立体异构体,确证了Callyspongiolide天然产物的立体结构,并结合抗癌活性实验,确证了Callyspongiolide天然产物以及它的三个立体异构体的优良的抗癌活性,具有很强的医药工业的应用前景。
权利要求
1.具有通式(I)所示结构的化合物及其药学上可接受的盐:其中,R1-R6独立的选自H、C1-C6的烷基,C1-C6的烷氧基,卤素,羟基,氨基,硝基,氰基,巯基
2.根据权利要求1所述的化合物,其特征在于:优选的,其中R1-R6为H或C1-C3的烷基。
3.根据权利要求2所述的化合物,其特征在于:优选的,其中R1-R3为甲基,R4-R6为H。
4.根据权利要求1-3所述的化合物,其特征在于:优选的,所述化合物的结构如下:
5.一种化合物1a和1b的合成方法,其特征在于,包括由化合物2与化合物3a或3b通过Sonogashira偶联反应制备的步骤,其中的X表示卤素取代基,Pg1和Pg2各自独立的选自常规的羟基保护基,进一步的,Pg1优选TES,Pg2优选TBS
6.一种化合物1c和1d的合成方法,其特征在于,包括由化合物ent-2与化合物3a或3b通过Sonogashira偶联反应制备的步骤,其中的X表示卤素取代基,Pg1和Pg2各自独立的选自常规的羟基保护基,进一步的,Pg1优选TES,Pg2优选TBS
7.根据权利要求5-6任一项所述的合成方法,其特征在于:所述偶联反应后进一步进行氨基甲酸酯化反应和脱除羟基保护基,从而获得终产物。
8.包含权利要求1-4任一项所述的化合物或其药学上可接受的盐的药物组合物。
9.权利要求1-4任一项所述的化合物或其药学上可接受的盐在制备治疗抗肿瘤药物中的用途。
10.具有如下结构的中间体化合物:
说明书
技术领域
本发明涉及大环内酯Callyspongiolide立体异构体的合成及抗癌应用,属于有机与药物化学领域。
背景技术
Callyspongiolide是从印度尼西亚海域的海绵Callyspongiasp.中分离得到的大环内酯类天然化合物(C.-D.Pham,R.Hartmann,P. B.Stork,S.Wesselborg,W.Lin,D.Lai,P.Proksch,Org.Lett.2014,16,266.),该分子含有两个主要的结构单元:14-元大环内酯结构片段和芳香苄醇结构片段,两者由中间的烯-炔-烯共轭体系连接。该分子在分离后的结构鉴定过程中尚未完成其绝对立体化学的确认,仅通过核磁共振(NMR)技术确定了大环内酯结构单元的相对立体化学和整个分子的碳骨架结构,绝对立体化学(包括右侧苄醇)并未确定下来。在针对该分子的生物学研究中,分离天然产物的作者确认Callyspongiolide对于部分的肿瘤细胞具有体外细胞毒性活性。
可见,进一步的确证天然产物Callyspongiolide的结构是亟待解决的技术问题,而基于Callyspongiolide的部分抗肿瘤活性的研究结果,结合不同肿瘤的发病机理各异,进一步的发现它更为广泛的抗肿瘤的应用也引起了人们的兴趣,而除此以外,基于Callyspongiolide结构的复杂性,可想而知其具有众多的立体异构体,技术人员们普遍知晓的是不同的立体异构体之间的生物活性往往千差万别,那么,研究发现更多的天然产物立体异构体,并确证他们的抗肿瘤活性,也将对于构效关系的研究以及抗癌候选药物的储备具有非常重要的价值。
因此,基于上述描述,确证天然产物Callyspongiolide的结构,并合成得到其立体异构体,表征它们的抗癌活性,仍是目前的研究热点和重点,这也正是本发明得以完成的基础所在和动力所倚。
发明内容
如上所述,为了确证天然产物Callyspongiolide的结构,并合成得到其立体异构体,表征它们的抗癌活性,本发明人对此进行了深入的研究,在付出大量创造性劳动后,从而完成了本发明。
本发明涉及如下四个方面,更具体而言,第一个方面,本发明涉及具有如下式(I)所示结构的化合物及其药学上可接受的盐,其中,R1-R6独立的选自H、C1-C6的烷基,C1-C6的烷氧基,卤素,羟基,氨基,硝基,氰基,巯基。
在本发明的所述化合物中,除非另有规定,卤素或卤代中的卤素例如可为F、Cl、Br或I。
在本发明的所述化合物中,C1-C6的烷基的含义是指具有1-6个碳原子的直链或支链烷基,非限定性地例如可为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、正戊基、异戊基或正己基等。
在本发明的所述化合物中,C1-C6烷氧基是指上述定义的“C1-C6烷基”与O原子相连后的基团。
优选的所述式(I)所示化合物的结构如下:
第二个方面,本发明涉及上述化合物的合成方法。
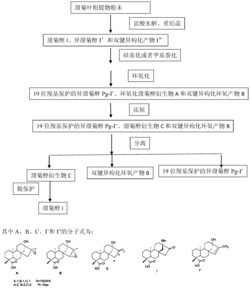
1、化合物1a和1b的合成
化合物1a/b的逆合成分析如下:
由于天然产物C-21位羟基的绝对构型未定,我们决定合成两个构型1a/b来确定其构型。从其特有的烯-炔-烯结构分割,化合物1a/b可以通过Sonogashira偶联反应将大环化合物2和溴代苯基化合物3a/b连接来得到。Yamaguchiesterification关大环将化合物4转化为化合物2。而3a/b中未定手性中心由Mukaiyamaaldol反应从化合物5来合成。化合物4中的顺式双键和反式双键分别由HWEreaction和Julia-Lythgoeolefination来构建。最后,化合物6由Krischeallylation转化合成,化合物7则由Marshall的TMSAcetylene的不对称加成获得。
所述化合物1a和1b的合成方法包括:由化合物2与化合物3a或3b通过Sonogashira偶联反应制备,所述偶联反应后进一步进行氨基甲酸酯化反应和脱除羟基保护基,从而获得终产物。
优选的,所述Sonogashira偶联反应在Pd催化剂(优选为Pd(PPh3)4),铜催化剂(优选为CuI)以及碱(优选为三乙胺)的存在下进行,所述氨基甲酸酯化反应在三氯乙酰异氰酸酯的存在下进行,所述脱除羟基保护基反应在TSAF的存在下进行。
进一步的,所述化合物2的制备方法包括:将化合物4经过Yamaguchiesterification关环反应实现制备。
优选的,化合物4首先脱除PMB,然后水解得到前体化合物,之后在三氯苯甲酰氯的存在下碱性条件下关环,并脱除保护基从而得到化合物2。
进一步的,所述化合物4的制备方法包括:将化合物6和7经过HWEreaction和Julia-Lythgoeolefination反应来制备。
进一步的,所述化合物6的制备方法包括:由化合物8经过Krischeallylation反应转化合成,所述化合物7的制备方法包括:由化合物9经过TMSAcetylene的不对称加成制备。
进一步的,所述化合物3a或3b的制备方法包括:由化合物5经过Mukaiyamaaldol反应制备。
具体合成路线如下:
1)片段6的合成
反应式1:片段6的合成
片段6的合成如上所示。化合物10经过立体控制的Krischeallyation由化合物8来构建。化合物10经过DDQ脱PMB、TBS保护二醇和硼氢化氧化双键三步反应得到11。化合物11一步氧化羟基到酸后,经过混酐条件接上Evens辅基12得到13。最后,经过辅基立体控制的甲基化反应和硼氢化锂还原去除辅基得得到片段6。
2)片段7的合成
反应式2:片段7的合成
片段7的合成如上所示。从简单化合物9出发,Marshall的TMSAcetylene的不对称加成可以得到立体专一的化合物14。用PMB基团保护所得羟基,碱性条件脱除TMS可以获得化合物15。钯催化的烯基碘化反应生成化合物16。通过TBAF条件脱除TBS,所得醇经过Mitsnunobu反应得到化合物18。最后,钼酸铵催化的氧化反应生成化合物7。
3)片段2的合成
反应式3:片段2的合成
DMP氧化将醇6转化为醛19。经过Julia-Lythgoeolefination与化合物7可以以E/Z=6:1的比例合成双键,CSA条件下选择性脱一级TBS得到20。DMP氧化可以获得醛,分子间的HWE反应可以以很好的产率单一的得到Z式构型的化合物4。DDQ脱PMB,水解乙酯可以单一地得到关环前体22。Yamaguchi大环内酯化后,CSA脱除二级TBS可以获得化合物2。
4)片段3a/b的合成
反应式4:片段3a/b的合成
片段3a/b的合成如上所示,经过TBS保护溴代苯甲醛5上的酚羟基,立体控制的Mukaiyamaaldol反应可以选择性的获得化合物24a/b。TES保护所得羟基和DIBAL还原甲酯可以合成相应的醇25a/b。所得羟基被DMP氧化成醛,紧接着醛进行Takai反应获得对应的烯基碘化合物,最后经过碱性后处理可以得到化合物26a/b。Sonogashira偶联反应生成相应的烯炔化合物,TBS保护裸露的酚羟基最终获得化合物3a/b。
5)片段连接
反应式5:片段连接
将已经获得的大环化合物2和溴代苯化合物3a/b通过Sonogashira偶联反应连接两个片段获得相应的天然产物前体。最后,氨基甲酸酯化反应和TASF脱除TES保护剂可以获得终产物1a和1b。
2、化合物1c和1d的合成
进一步的利用与上述相似的工艺具体的替换了反应原料合成了1c和1d,逆合成分析如下,具体不再重复赘述。
具体的,所述化合物1c和1d的合成方法包括:由化合物ent-2与化合物3a或3b通过Sonogashira偶联反应制备。
优选的,所述Sonogashira偶联反应在Pd催化剂(优选为Pd(PPh3)4),铜催化剂(优选为CuI)以及碱(优选为三乙胺)的存在下进行,所述氨基甲酸酯化反应在三氯乙酰异氰酸酯的存在下进行,所述脱除羟基保护基反应在TSAF的存在下进行。
进一步的,所述化合物ent-2的制备方法包括:将化合物4经过Yamaguchiesterification关环反应实现制备。
优选的,化合物4首先脱除PMB,然后水解得到前体化合物,之后在三氯苯甲酰氯的存在下碱性条件下关环,并脱除保护基从而得到化合物2。
进一步的,所述化合物4的制备方法包括:将化合物5和6经过HWEreaction和Julia-Lythgoeolefination反应来制备。
进一步的,所述与前述结构相同的中间体化合物3a或3b的制备方法也同前面描述包括:由化合物2-溴3-羟基苯甲醛经过Mukaiyamaaldol反应制备。
具体的片段构建方法如下:
反应式6:片段5的合成
反应式7:片段6的合成
反应式8:片段ent-2的合成
反应式9:片段的连接
第三个方面,本发明涉及上述式(I)化合物和/或其药学上可接受的盐在制备治疗抗肿瘤药物中的用途。尤其涉及化合物(1a、1b、1c、1d)和/或其药学上可接受的盐在制备治疗抗肿瘤药物中的用途。所述肿瘤包括但不限于乳腺癌、成神经细胞瘤、胃癌、肝癌、肺癌、结肠癌、宫颈癌、前列腺癌、白血病。
第四方面,本发明涉及上述式(I)化合物和/或其药学上可接受的盐的药物组合物。尤其涉及化合物(1a、1b、1c、1d)和/或其药学上可接受的盐与药学上可接受的载体和/或其它抗肿瘤药物组成的药物组合物。所述药物组合物可以采用有机领域公知的技术制备。
作为一种例举性的药物组合物的制备方法如下:使本发明化合物与制剂学上可接受的固体或液体载体结合,以及使之任意地与制剂学上可接受的辅助剂和赋形剂结合制备成微粒或微球。固体剂型包括片剂、分散颗粒、胶囊、缓释片、缓释微丸等等。固体载体可以是至少一种物质,其可以充当稀释剂、香味剂、增溶剂、润滑剂、悬浮剂、粘合剂、崩解剂以及包裹剂。惰性固体载体包括磷酸镁、硬脂酸镁、滑粉糖、乳糖、果胶、丙二醇、聚山梨酯80、糊精、淀粉、明胶、纤维素类物质例如甲基纤维素、微晶纤维素、低熔点石蜡、聚乙二醇、甘露醇、可可脂等。液体剂型包括溶剂、悬浮液例如注射剂、粉剂等等。
药物组合物以及单元剂型中含有的活性成份(本发明化合物)的量可以根据患者的病情、医生诊断的情况特定地加以应用,所用的化合物的量或浓度在一个较宽的范围内调节,通常,活性化合物的量范围为组合物的0.5%-90%(重量)。另一优选的范围为0.5%-70%。
附图说明
图1是化合物1a在抑制癌症细胞株的增殖方面的效果。
图2是化合物1b在抑制癌症细胞株的增殖方面的效果。
图3是化合物1c在抑制癌症细胞株的增殖方面的效果。
图4是化合物1d在抑制癌症细胞株的增殖方面的效果。
具体实施方式
下面通过具体的实施例对本发明进行详细说明,但这些例举性实施方式的用途和目的仅用来例举本发明,并非对本发明的实际保护范围构成任何形式的任何限定,更非将本发明的保护范围局限于此。
实施例1
氮气保护下,将醇8(200mg,0.84mmol),10(44mg,0.04mmol),Cs2CO3(85mg,0.50mmol),m-ClBzOH(13mg,0.08mmol)和H2O(30mg,1.68mmol)加入封管中,随后加入乙酸烯丙酯(168mg,1.68mmol)。体系在100℃反应20小时后,减压将低沸点溶液旋走。剩余物质经过硅胶柱分离得到无色油状物产品11(210mg,90%)。Rf=0.2(硅胶,10%乙酸乙酯的正己烷溶液);[α]D20=-9.4(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ7.27(d,J=8.2Hz,2H),6.89(d,J=8.6Hz,2H),5.89–5.76(m,1H),5.14(dd,J=7.7,6.8Hz,2H),4.44(s,2H),3.81(s,3H),3.79–3.69(m,1H),3.53–3.43(m,2H),2.33–2.22(m,1H),2.18–2.07(m,1H),1.91(s,1H),1.83–1.60(m,3H),1.46–1.31(m,3H),0.94(d,J=6.6Hz,3H).13CNMR(125MHz,CDCl3)δ159.23,134.96,130.69,129.27,117.89,113.84,72.67,68.62,68.33,55.29,44.34,42.20,36.26,27.02,20.69.HRMS(m/z):C17H26O3Na+[M+Na]+:301.1774,301.1775.
实施例2
11(300mg,1.1mmol)溶解于DCM(10mL)和pH7.0的缓冲溶液(1mL)中。随后DDQ(500mg,2.2mmol)加入。体系室温反应2小时。最后,反应体系逐次由饱和的Na2S2O3溶液(20mL),饱和的NaHCO3溶液(20mL)和饱和食盐水(20mL)洗涤。收集有机相,并用无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物11-1(162mg,95%)。TLC:Rf=0.4(硅胶,33%乙酸乙酯的正己烷溶液);[α]D20=–22.0(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ5.86–5.75(m,1H),5.13–5.05(m,2H),3.78–3.71(m,1H),3.71–3.65(m,1H),3.65–3.56(m,1H),2.95(s,2H),2.29–2.19(m,1H),2.19–2.09(m,1H),1.88–1.73(m,1H),1.73–1.62(m,1H),1.42–1.31(m,2H),1.31–1.18(m,1H),0.92(d,J=6.7Hz,3H).13CNMR(125MHz,CDCl3)δ134.99,117.79,68.22,60.58,44.26,42.25,38.52,25.88,20.75.HRMS(m/z):C9H18O2Na+[M+Na]+:181.1199,181.1192.
实施例3
二醇11-1(272mg,1.74mmol)溶解在DCM(15mL)中。–78℃条件下,Et3N(0.95mL,7.00mmol)和TBSOTf(1.13mL,5.25mmol)先后缓慢滴加入反应体系中。随后体系升温至–30℃,并在–30℃下反应2小时。再将反应温度降到–78℃,饱和的NaHCO3溶液(20mL)猝灭反应。然后升至室温,分液。水相用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物11-2(670mg,99%)。TLC:Rf=0.20(硅胶,正己烷溶液);
11-2(670mg,1.76mmol)溶解于THF(10mL)中,并将温度降至0℃。随后9-BBN(12mL,0.5M)逐滴加入。缓慢升至室温,继续反应2h。0℃条件下,饱和NaHCO3(10mL)溶液和30%双氧水(10mL)缓慢加入。升至室温,继续反应2小时。分液,水相用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物12(630mg,90%)。Rf=0.4(硅胶,10%乙酸乙酯的正己烷溶液);[α]D20=1.8(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ3.88–3.79(m,1H),3.69–3.55(m,4H),2.24(s,1H),1.67–1.52(m,7H),1.37–1.23(m,2H),0.91–0.83(m,22H),0.07(s,6H),0.05(s,6H).13CNMR(125MHz,CDCl3)δ70.16,63.24,61.11,44.12,40.48,33.14,27.87,26.29,25.97,25.90,19.94,18.33,18.10,-4.43,-4.46,-5.26,-5.29.HRMS(m/z):C21H48O3Si2Na+[M+Na]+:427.3034,427.3036.
实施例4
醇12(630mg,1.56mmol)首先溶解于CH3CN(20mL)和pH7.0的缓冲溶液(20mL)中。随后NaClO2(755mg,8.39mmol),NaClO(1.55mL,10%availablechlorine)和TEMPO(15mg,0.10mmol)逐次加入,2小时后,用1MHCl溶液将反应体系调到pH3。减压旋走低沸点溶剂。水相用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物12-1(610mg,94%)。Rf=0.4(硅胶,10%乙酸乙酯的正己烷溶液);[α]D20=10.1(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ3.87–3.78(m,1H),3.71–3.58(m,2H),2.44(t,J=7.6Hz,2H),1.93–1.80(m,1H),1.70–1.59(m,2H),1.58–1.50(m,1H),1.46–1.39(m,1H),1.39–1.26(m,2H),0.94–0.89(m,12H),0.89(s,9H),0.06(s,6H),0.05(s,6H).13CNMR(125MHz,CDCl3)δ69.30,61.14,44.69,40.51,31.19,29.57,26.38,26.00,25.91,19.95,18.34,18.07,-4.31,-4.51,-5.26,-5.28.HRMS(m/z):C21H46O4Si2Na+[M+Na]+:441.2827,441.2825.
实施例5
酸12-1(610mg,1.46mmol)溶于无水THF(10mL)溶液,并降温至0℃。Et3N(0.61mL,4.38mmol)和特戊酰氯(180uL,1.61mmol)逐次缓慢滴加入反应体系。反应持续2小时后,反应体系降至-78℃。在另一个烧瓶中,正丁基锂(1.1mL,1.65mmol,1.5M)在-78℃条件下滴加入(S)-4-苄基-2-唑烷酮(550mg,3.11mmol)的THF(5mL)的溶液中。30分钟后,将此反应溶液加入到上面的反应体系中。-78℃条件反应3小时。饱和的NaHCO3溶液(20mL)猝灭反应。然后升至室温,分液。水相用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物13(675mg,80%)。Rf=0.9(硅胶,17%乙酸乙酯的正己烷溶液);[α]D20=35.4(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ7.34(t,J=7.2Hz,2H),7.29(d,J=7.3Hz,1H),7.24–7.18(m,2H),4.71–4.58(m,1H),4.22–4.12(m,2H),3.89–3.81(m,1H),3.71–3.57(m,2H),3.31(dd,J=13.4,3.2Hz,1H),3.08–2.91(m,2H),2.77(ddd,J=14.7,9.6,5.1Hz,1H),2.00–1.89(m,1H),1.76–1.63(m,2H),1.63–1.56(m,1H),1.47(ddd,J=13.1,7.5,5.4Hz,1H),1.36(ddd,J=13.8,8.0,6.1Hz,2H),0.93(d,J=6.6Hz,3H),0.90(s,18H),0.06(s,6H),0.05(s,6H).13CNMR(125MHz,CDCl3)δ173.52,153.40,135.40,129.46,128.99,127.36,69.41,66.16,61.31,55.19,45.24,40.54,37.96,31.63,30.96,26.48,26.03,25.95,20.05,18.37,18.09,-4.28,-4.48,-5.21,-5.24.HRMS(m/z):C31H55NO5Si2Na+[M+Na]+:600.3511,600.3512.
实施例6
-78℃条件下,将NaHMDS(0.85mL,1.70mmol,2.0M)加入到13(395mg,0.68mmol)的THF(10mL)溶液中。然后,逐滴缓慢加入MeI(131uL,2.05mmol)。反应体系搅拌过夜。饱和的NH4Cl溶液(3mL)猝灭反应。然后升至室温,分液。水相用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物13-1(420mg,99%)。Rf=0.80(硅胶,17%乙酸乙酯的正己烷溶液);[α]D20=49.1(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ7.34(t,J=7.2Hz,2H),7.29(d,J=7.3Hz,1H),7.25–7.20(m,2H),4.75–4.61(m,1H),4.27–4.05(m,2H),3.89–3.81(m,1H),3.81–3.72(m,1H),3.71–3.59(m,2H),3.25(dd,J=13.4,3.2Hz,1H),2.78(dd,J=13.4,9.6Hz,1H),2.15(ddd,J=13.6,9.6,3.7Hz,1H),1.73–1.64(m,1H),1.60–1.53(m,1H),1.52–1.45(m,1H),1.42–1.34(m,2H),1.26(d,J=7.0Hz,3H),0.94(d,J=6.6Hz,3H),0.91(s,9H),0.87(s,9H),0.07(s,6H),0.03(s,3H),-0.02(s,3H).13CNMR(125MHz,CDCl3)δ176.87,152.76,135.36,129.53,128.95,127.36,68.78,65.93,61.22,55.22,45.51,40.82,39.84,37.91,34.19,26.47,26.04,25.89,19.90,19.08,18.37,18.03,-4.13,-4.74,-5.21,-5.23.HRMS(m/z):C32H57NO5Si2Na+[M+Na]+:614.3667,614.3669.
实施例7
0℃条件下,将硼氢化锂(1.2mL,2.4mmol,2M)加入到13-1(420mg,0.71mmol)的THF(10mL)溶液中。随后,加入MeOH(0.2mL)。升至室温,继续反应3小时。体系降至0℃,饱和NaHCO3(10mL)淬灭反应,并继续搅拌2小时。减压旋干低沸点溶剂。水相用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物6(260mg,88%)。Rf=0.4(硅胶,10%乙酸乙酯的正己烷溶液);[α]D20=4.8(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ4.00–3.92(m,1H),3.76–3.69(m,1H),3.69–3.56(m,2H),3.47(s,1H),3.38–3.26(m,1H),1.97–1.84(m,1H),1.66–1.55(m,4H),1.49–1.38(m,1H),1.36–1.30(m,2H),0.91(s,9H),0.91–0.88(m,12H),0.87(d,J=6.8Hz,3H),0.11(s,3H),0.10(s,3H),0.05(s,6H).13CNMR(125MHz,CDCl3)δ69.80,68.80,60.91,43.37,41.99,40.58,31.57,26.21,25.98,25.86,19.65,18.80,18.28,18.09,-4.54,-4.58,-5.26,-5.29.HRMS(m/z):C22H50O3Si2Na+[M+Na]+:441.3191,441.3193.
实施例8
TMS乙炔(2.6mL,18.8mmol)缓慢加入Et2Zn(18.8mL,1.0M,18.8mmol)的甲苯溶液中。随后,反应体系加热回流1小时。降至室温后,(S)-BINOL(0.52g,1.88mmol)的Et2O(20mL)溶液和Ti(OiPr)4(1.39mL,4.70mmol)逐次加入。1小时后,醛9(0.95g,4.70mmol)的Et2O(10mL)溶液缓慢加入到反应液中。反应搅拌过夜。1M的酒石酸溶液(50mL)淬灭反应。搅拌30分钟后,分离有机相。水相用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物14(1.14g,80%)。Rf=0.6(硅胶,10%乙酸乙酯/己烷);[α]D20=-6.2(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ4.39(t,J=5.8Hz,1H),3.94(dd,J=10.0,4.0Hz,1H),3.57(dd,J=10.0,6.6Hz,1H),3.46(d,J=5.4Hz,1H),1.93(ddd,J=13.3,6.7,4.1Hz,1H),1.60(s,1H),1.02(d,J=7.0Hz,3H),0.90(s,9H),0.18(s,9H),0.08(s,3H),0.08(s,3H).13CNMR(125MHz,CDCl3)δ105.79,89.84,67.10,66.73,40.59,25.83,18.18,12.88,-0.11,-5.59,-5.64.HRMS(m/z):C15H32O2Si2Na+[M+Na]+:323.1833,323.1839.
实施例9
0℃条件下,将NaH(27mg,0.67mmol,60%)加入到化合物14(100mg,0.33mmol)的THF(5mL)溶液中。30分钟后,卞溴(59.6uL,0.40mmol)在0℃条件下加入。反应升至室温,并搅拌过夜。饱和的NH4Cl溶液(3mL)猝灭反应。用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物15(85mg,73%)。TLC:Rf=0.7(硅胶,10%乙酸乙酯的正己烷溶液);[α]D20=41.4(c0.5,CHCl3);1HNMR(500MHz,CDCl3)δ7.29(d,J=8.6Hz,2H),6.88(d,J=8.7Hz,2H),4.76(d,J=11.4Hz,1H),4.45(d,J=11.4Hz,1H),4.21(dd,J=6.0,2.1Hz,1H),3.81(s,3H),3.63–3.47(m,2H),2.46(d,J=2.1Hz,1H),2.03(dt,J=12.5,6.3Hz,1H),1.01(d,J=6.8Hz,3H),0.86(s,9H),0.03(s,6H).13CNMR(125MHz,CDCl3)δ159.27,130.16,129.58,113.80,81.50,74.73,70.47,69.80,64.39,55.29,40.47,25.89,18.25,12.22,-5.41,-5.48.HRMS(m/z):C20H32NaO3SNa+[M+Na]+:371.2013,371.2005.
实施例10
化合物15(85mg,0.24mmol)溶解于THF(5mL)中。随后逐次加入Pd(PPh3)4(14mg,0.012mmol)和n-Bu3SnH(79uL,0.29mmol)。20分钟后,减压出去THF。剩余物质快速硅胶柱分离得到无色油状物质。将此物质溶于DCM(5mL)中。I2(1M的CH2Cl2溶液)逐滴加入到反应体系中,直到反应液由无色变为浅红色且半分钟内不变色。饱和的Na2SO3溶液(3mL)猝灭反应。用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物16(90mg,77%)。TLC:Rf=0.80(硅胶,5%乙酸乙酯的正己烷溶液);[α]D20=9.7(c1.0,CHCl3);1HNMR(400MHz,CDCl3)δ7.24(d,J=8.6Hz,2H),6.88(d,J=8.6Hz,2H),6.49(dd,J=14.5,8.2Hz,1H),6.27(d,J=14.5Hz,1H),4.51(d,J=11.3Hz,1H),4.28(d,J=11.4Hz,1H),3.82–3.77(m,4H),3.64–3.50(m,2H),1.87(dt,J=12.3,5.6Hz,1H),0.90–0.87(m,12H),0.03(s,6H).13CNMR(100MHz,CDCl3)δ159.13,145.35,130.41,129.27,113.78,82.27,78.42,70.40,64.24,55.28,39.87,25.92,18.25,12.64,-5.41,-5.44.HRMS(m/z):C20H33IO3SiNa+[M+Na]+:499.1136,499.1137.
实施例11
TBAF(1.5mL,1.50mmol,1.0M的THF溶液)滴加入稀基碘16(335mg,0.70mmol)的THF(10mL)溶液中。反应体系在室温条件下搅拌3小时。饱和的NH4Cl溶液(3mL)猝灭反应。用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物16-1(250mg,98%)。TLC:Rf=0.20(硅胶,33%乙酸乙酯的正己烷溶液);[α]D20=45.0(c1.0,CHCl3);1HNMR(400MHz,CDCl3)δ7.23(d,J=8.6Hz,2H),6.89(d,J=8.7Hz,2H),6.48(dd,J=14.6,8.3Hz,1H),6.34(d,J=14.6Hz,1H),4.56(d,J=11.4Hz,1H),4.27(d,J=11.4Hz,1H),3.81(s,3H),3.72–3.57(m,2H),3.58–3.47(m,1H),1.87(ddd,J=14.9,7.1,3.7Hz,1H),0.83(d,J=7.0Hz,3H).13CNMR(100MHz,CDCl3)δ159.41,145.38,129.56,129.53,114.01,86.06,79.59,70.44,66.58,55.30,39.49,13.51.HRMS(m/z):C14H19IO3Na+[M+Na]+:385.0271,385.0276.
实施例12
醇16-1(250mg,0.69mmol)溶于THF(20mL)溶液中。反应体系降至0℃,5-疏基-1-苯基-四氮唑(200mg,1.12mmol),Ph3P(300mg,1.12mmol),和DEAD(170uL,1.12mmol)逐次加入到反应体系中。反应搅拌2小时。饱和的NaHCO3溶液(20mL)猝灭反应。用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物18(328mg,91%)。TLC:Rf=0.40(硅胶,33%乙酸乙酯的正己烷溶液);[α]D20=10.0(c1.0,CHCl3);1HNMR(400MHz,CDCl3)δ7.57–7.49(m,5H),7.22(d,J=8.6Hz,2H),6.84(d,J=8.6Hz,2H),6.48(dd,J=14.6,7.9Hz,1H),6.38(d,J=14.6Hz,1H),4.54(d,J=11.4Hz,1H),4.26(d,J=11.4Hz,1H),3.79(s,3H),3.70–3.55(m,2H),3.46–3.36(m,1H),2.21–2.16(m,1H),1.02(d,J=6.9Hz,3H).13CNMR(100MHz,CDCl3)δ159.28,154.63,144.51,133.73,130.07,129.78,129.68,129.58,123.86,113.83,113.74,83.67,79.95,70.39,55.27,37.32,36.52,15.45.HRMS(m/z):C21H23IN4O2SNa+[M+Na]+:545.0479,545.0477.
实施例13
18(51mg,0.10mmol)溶于EtOH(10mL)溶液中。随后,(NH4)6Mo7O24·4H2O(50mg,0.04mmol)和30%H2O2(2mL)逐次加入。反应室温搅拌24小时。用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物7(45.3mg,84%)。TLC:Rf=0.39(硅胶,33%乙酸乙酯的正己烷溶液);[α]D20=20.4(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ7.67–7.51(m,5H),7.21(d,J=8.6Hz,2H),6.89(d,J=8.6Hz,2H),6.50–6.36(m,2H),4.54(d,J=11.4Hz,1H),4.26(d,J=11.4Hz,1H),4.07(dd,J=14.6,3.6Hz,1H),3.82(s,3H),3.73–3.63(m,1H),3.53(dd,J=14.6,8.8Hz,1H),1.16(d,J=6.9Hz,3H).13CNMR(125MHz,CDCl3)δ159.47,154.05,143.58,133.08,131.52,129.69,129.59,129.35,125.30,114.01,83.19,81.01,70.60,58.23,55.35,32.80,16.22.HRMS(m/z):C21H23IN4O4SNa+[M+Na]+:577.0377,577.0378.
实施例14
0℃条件下,NaHCO3(50mg,0.60mmol)和Dess-Martinperiodinane(250mg,0.59mmol)逐次加入到化合物6(150mg,0.36mmol)的DCM(5mL)溶液中。反应继续1小时后,减压旋干直接硅胶柱分离得到无色油状物19(140mg,94%),直接用于下一步。
化合物7(300mg,0.54mmol)溶于THF(3mL)中,并降温到-78℃。LHMDS(0.56ml,1M)逐滴加入。搅拌30分钟后,醛19(140mg,0.34mmol)逐滴加入。反应30分钟后,体系升至室温。搅拌过夜。饱和的NH4Cl溶液(20mL)猝灭反应。用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物20(190mg,76%,C10/11E/Z=6:1)。TLC:Rf=0.70(硅胶,5%乙酸乙酯的正己烷溶液);[α]D20=27.6(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ7.24(d,J=8.5Hz,2H),6.88(d,J=8.6Hz,2H),6.47(dd,J=14.5,7.9Hz,1H),6.24(d,J=14.5Hz,1H),5.42–5.25(m,2H),4.52(d,J=11.6Hz,1H),4.29(d,J=11.6Hz,1H),3.82(s,3H),3.74–3.69(m,1H),3.67–3.62(m,2H),3.59–3.53(m,1H),2.41–2.25(m,2H),1.67–1.61(m,1H),1.59–1.54(m,1H),1.46–1.39(m,2H),1.37–1.31(m,2H),1.30–1.27(m,1H),0.98(m,6H),0.92–0.89(m,21H),0.06(s,12H).13CNMR(125MHz,CDCl3)δ159.17,145.30,137.03,130.45,129.82,129.19,113.78,85.03,78.24,70.22,69.06,61.26,55.29,45.76,44.85,40.79,40.57,33.00,26.37,26.04,21.90,20.24,18.36,18.11,15.95,-3.82,-4.02,-5.22,-5.23.HRMS(m/z):C36H65IO4Si2Na+[M+Na]+:767.3358,767.3353.
实施例15
20(100mg,0.13mmol)溶于MeOH(5mL)中,并降至0℃。CSA(10mg,0.04mmol)加入体系。0℃条件下,反应3小时。Et3N(5uL,0.04mmol)淬灭反应。减压旋干低沸点溶剂。硅胶柱分离获得无色油状物21(83mg,98%,C10/11E/Z=6:1)。TLC:Rf=0.40(硅胶,10%乙酸乙酯的正己烷溶液);[α]D20=32.3(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ7.24(d,J=8.4Hz,2H),6.88(d,J=8.4Hz,2H),6.52–6.39(m,1H),6.24(d,J=14.5Hz,1H),5.40–5.24(m,2H),4.52(d,J=11.6Hz,1H),4.29(d,J=11.6Hz,1H),3.81(s,3H),3.76–3.72(m,1H),3.71–3.65(m,2H),3.59–3.47(m,1H),2.39–2.22(m,2H),1.71–1.60(m,3H),1.43–1.33(m,4H),0.98(m,6H),0.89(m,12H),0.06(s,6H).13CNMR(125MHz,CDCl3)δ159.14,145.30,136.96,130.40,129.88,129.23,113.78,84.96,78.37,77.33,77.08,76.82,70.18,69.01,60.98,55.31,45.61,44.74,40.81,40.41,33.08,26.04,21.89,20.22,18.13,16.09,-3.83,-4.00.HRMS(m/z):C30H51IO4SiNa+[M+Na]+:653.2494,653.2490.
实施例16
0℃条件下,NaHCO3(69mg,0.82mmol)和Dess-Martinperiodinane(150mg,0.35mmol)逐次加入到化合物6(150mg,0.24mmol)的DCM(5mL)溶液中。反应继续1小时后,减压旋干直接硅胶柱分离得到无色油状物(140mg,94%),直接用于下一步。
KHMDS(714uL,0.36mmol,0.5M)在-78℃的条件下缓慢加入18-crown-6(100mg,0.36mmol)和(CF3CH2)P(O)CH2CO2Me(114mg,0.36mmol)的THF(7mL)体系中。15分钟后,上步反应的醛的THF溶液逐滴加入到反应体系中。-78℃的条件下反应过夜。饱和NH4Cl溶液(20mL)猝灭反应。用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物23(147mg,96%,C10/11E/Z=6:1)。LC:Rf=0.65(硅胶,10%乙酸乙酯的正己烷溶液);[α]D20=26.3(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ7.23(d,J=8.5Hz,2H),6.88(d,J=8.5Hz,2H),6.46(dd,J=14.5,7.9Hz,1H),6.26–6.18(m,2H),5.84(d,J=11.6Hz,1H),5.41–5.22(m,2H),4.52(d,J=11.6Hz,1H),4.29(d,J=11.6Hz,1H),3.81(s,3H),3.76–3.69(m,4H),3.60–3.52(m,1H),2.65–2.57(m,2H),2.38–2.31(m,1H),2.31–2.22(m,1H),1.74–1.65(m,1H),1.50–1.29(m,4H),1.02–0.94(m,6H),0.91–0.87(m,12H),0.06(s,3H),0.05(s,3H).13CNMR(125MHz,CDCl3)δ166.86,159.18,149.33,145.33,136.97,130.44,129.83,129.19,120.22,113.78,85.00,78.25,70.22,68.91,55.29,50.96,45.06,44.93,40.80,36.26,33.01,29.83,26.01,21.74,20.09,18.11,16.00,-3.93,-4.01.HRMS(m/z):C33H53IO5Na+[M+Na]+:707.2599,707.2593.
实施例17
23(110mg,0.17mmol)溶解于DCM(7mL)和pH7.0的缓冲溶液(1mL)中。随后DDQ(120mg,0.53mmol)加入。体系室温反应2小时。最后,反应体系逐次由饱和的Na2S2O3溶液(20mL),饱和的NaHCO3溶液(20mL)和饱和食盐水(20mL)洗涤。收集有机相,并用无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物24(74mg,82%).TLC:Rf=0.40(硅胶,5%乙酸乙酯的正己烷溶液);[α]D20=13.0(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ6.56(dd,J=14.4,6.6Hz,1H),6.36(d,J=14.5Hz,1H),6.28–6.19(m,1H),5.84(d,J=11.6Hz,1H),5.46–5.21(m,2H),3.83–3.73(m,2H),3.71(s,3H),2.61(t,J=6.5Hz,2H),2.37–2.30(m,1H),2.25–2.14(m,1H),1.71–1.58(m,2H),1.46–1.34(m,4H),1.01–0.96(m,6H),0.91–0.87(m,12H),0.06(s,3H),0.05(s,3H)少1H.13CNMR(125MHz,CDCl3)δ166.88,149.31,146.70,139.54,129.34,120.24,77.93,69.02,50.99,44.99,44.82,43.19,36.12,33.28,29.86,29.71,26.00,21.85,20.05,18.14,16.33,-3.92,-4.02.HRMS(m/z):C25H45IO4Na+[M+Na]+:587.2024,587.2023.
实施例18
24(74mg,0.13mmol)溶于THF(5mL),MeOH(1mL)和H2O(3mL)的混合体系中。随后0℃条件下,LiOH(20mg,0.49mmol)加入。体系缓慢升至室温,继续搅拌5小时。饱和NH4Cl溶液(20mL)猝灭反应。用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物4(62mg,86%)。TLC:Rf=0.50(硅胶,25%乙酸乙酯/正己烷);[α]D20=20.6(c0.35,CHCl3);1HNMR(400MHz,CDCl3)δ6.55(dd,J=14.4,6.6Hz,1H),6.40–6.26(m,2H),5.86(d,J=11.6Hz,1H),5.50–5.17(m,2H),3.86–3.71(m,2H),2.70–2.53(m,2H),2.38–2.25(m,1H),2.25–2.13(m,1H),1.78–1.65(m,1H),1.49–1.32(m,4H),1.02–0.94(m,6H),0.94–0.87(m,12H),0.06(s,3H),0.05(s,3H).13CNMR(100MHz,CDCl3)δ170.13,151.53,146.49,139.67,129.29,119.85,78.00,69.01,44.93,44.80,43.19,36.12,33.30,29.93,29.71,25.99,21.93,20.12,18.14,16.46,-3.92,-4.05.HRMS(m/z):C24H43IO4Na+[M+Na]+:573.1868,573.1868.
实施例19
0℃条件下,2,4,6-trichlorobenzoylchloride(95μL,0.09mmol)逐滴加入到4(15mg,0.03mmol)和Et3N(18μL,0.12mmol)的体系中。反应升至室温,并搅拌1小时。随后,DMAP(18mg,0.15mmol)的甲苯(5mL)溶液加入。反应体系搅拌过夜。饱和NH4Cl溶液(20mL)猝灭反应。用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物,直接用于下一步。
上步所得化合物溶于MeOH(5mL)中,并降至0℃。CSA(2mg,0.01mmol)加入体系。升至室温条件下,反应3小时。Et3N(1.2uL,0.01mmol)淬灭反应。减压旋干低沸点溶剂。硅胶柱分离获得无色油状物2(7.6mg,67%,twosteps)。TLC:Rf=0.40(硅胶,10%乙酸乙酯的正己烷溶液);[α]D20=90.0(c0.2,CHCl3);1HNMR(400MHz,CDCl3)δ6.56–6.43(m,2H),6.34–6.23(m,1H),5.87(dd,J=11.6,2.5Hz,1H),5.18–5.10(m,2H),5.03(dd,J=15.0,9.2Hz,1H),3.81–3.70(m,1H),3.37(t,J=10.7Hz,1H),2.37–2.26(m,1H),2.25–2.09(m,2H),1.96(dd,J=14.8,2.8Hz,1H),1.68–1.56(m,2H),1.39–1.26(m,3H),1.05(d,J=7.1Hz,3H),0.98–0.93(m,6H).13CNMR(100MHz,CDCl3)δ165.08,146.50,143.13,137.75,131.56,121.64,81.05,65.95,46.00,42.98,42.68,34.49,31.24,29.71,27.09,22.35,20.04,17.53.HRMS(m/z):C18H27IO3Na+[M+Na]+:441.0897,441.0893.
实施例20
0℃条件下,咪唑(500mg,7.43mmol)和TBSCl(600mg,4.03mmol)逐次加入到5(500mg,2.25mmol)的DCM(20mL)中。反应升至室温,继续反应1小时。饱和NH4Cl溶液(20mL)猝灭反应。用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物5-1(775mg,99%)。TLC:Rf=0.85(硅胶,10%乙酸乙酯的正己烷溶液);
0℃条件下,BH3.THF(2.5mL,1.0M)加入到N-Ts-D-Val(676mg,2.46mmol)的DCM(7mL)溶液中。搅拌半个小时后,升至室温并搅拌1小时。随后体系降温至-78℃,5-1(775mg,2.46mmol)的DCM(3mL)溶液和25(0.73mL,3.20mmol)缓慢加入。-78℃条件下,反应12小时。饱和NH4Cl溶液(20mL)猝灭反应。用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物26(1.0g,98%)。TLC:Rf=0.45(硅胶,20%乙酸乙酯的正己烷溶液);[α]D20=20.8(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ7.18(t,J=7.9Hz,1H),7.09(dd,J=7.8,1.4Hz,1H),6.84(dd,J=7.9,1.5Hz,1H),5.57(d,J=4.6Hz,1H),3.76(s,3H),3.42(d,J=4.7Hz,1H),1.23(s,3H),1.21(s,3H),1.05(s,9H),0.25(s,3H),0.24(s,3H).13CNMR(125MHz,CDCl3)δ178.45,152.34,141.43,127.26,121.87,119.23,118.14,52.26,48.61,25.84,23.58,19.06,18.45,-4.10,-4.21.HRMS(m/z):C18H29BrO4SiNa+[M+Na]+:439.0911,439.0910.
实施例21
26(680mg,1.63mmol)溶解在DCM(15mL)中。–50℃条件下,Et3N(0.70mL,5.03mmol)和TESOTf(0.60mL,2.66mmol)先后缓慢滴加入反应体系中。随后体系升温至–30℃,并在–30℃下反应2小时。再将反应温度降到–50℃,饱和的NaHCO3溶液(20mL)猝灭反应。分液后水相用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物26-1(848mg,98%)。TLC:Rf=0.90(硅胶,20%乙酸乙酯的正己烷溶液);[α]D20=-10.8(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ7.18–7.09(m,2H),6.83(dd,J=5.6,3.9Hz,1H),5.70(s,1H),3.72(s,3H),1.20(s,3H),1.08(s,3H),1.06(s,9H),0.81(t,J=7.9Hz,9H),0.47–0.38(m,6H),0.24(s,3H),0.24(s,3H).13CNMR(125MHz,CDCl3)δ177.20,151.99,142.23,126.62,123.66,119.27,117.92,51.82,50.09,25.91,22.93,18.49,18.23,6.66,4.66,-4.16,-4.24.HRMS(m/z):C24H43BrO4Si2Na+[M+Na]+:553.1775,553.1774.
实施例22
DIBAL-H(2mL,1.5M)在-78℃条件加入26-1(548mg,1.03mmol)的DCM(15mL)溶液中。随后,反应体系温度升到-40℃,并反应1小时。-78℃条件下,MeOH(1mL)淬灭反应。升至室温,饱和酒石酸钾钠(20mL)加入并搅拌至澄清。分液后水相用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物27(485mg,95%)。TLC:Rf=0.20(硅胶,3%乙酸乙酯的正己烷溶液);[α]D20=2.6(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ7.24–7.14(m,2H),6.84(dd,J=7.6,1.9Hz,1H),5.26(s,1H),3.75(dd,J=11.0,3.5Hz,1H),3.52(dd,J=6.9,3.7Hz,1H),3.28(dd,J=11.0,6.9Hz,1H),1.16(s,3H),1.05(s,9H),0.85(t,J=7.9Hz,9H),0.76(s,3H),0.56–0.42(m,6H),0.24(s,3H),0.23(s,3H).13CNMR(125MHz,CDCl3)δ152.12,142.74,127.08,122.83,119.23,117.81,80.80,70.51,40.49,25.91,23.42,20.52,18.49,6.65,4.56,-4.14,-4.25.HRMS(m/z):C23H43BrO3Si2Na+[M+Na]+:525.1826,525.1827.
实施例23
0℃条件下,NaHCO3(200mg,2.38mmol)和Dess-Martinperiodinane(500mg,1.2mmol)逐次加入到化合物27(210mg,0.56mmol)的DCM(10mL)溶液中。反应继续1小时后,减压旋干直接硅胶柱分离得到无色油状物,直接用于下一步。
上步所得醛和碘仿(616mg,1.76mmol)加入到CrCl2(510mg,4.15mmol)的THF(5mL)溶液中。反应搅拌过夜。饱和的NaHCO3溶液(20mL)猝灭反应。分液后水相用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物28(145mg,68%)。TLC:Rf=0.20(硅胶,3%乙酸乙酯的正己烷溶液);[α]D20=52.3(c1.0,CHCl3);1HNMR(500MHz,CDCl3)δ7.19(t,J=7.9Hz,1H),7.04–6.89(m,2H),6.72(d,J=14.7Hz,1H),5.88(d,J=14.7Hz,1H),5.64(s,1H),4.89(s,1H),1.08(s,3H),1.01(s,3H),0.85(t,J=7.9Hz,9H),0.55–0.40(m,6H).13CNMR(125MHz,CDCl3)δ152.32,151.41,141.86,127.56,122.39,114.76,112.35,78.81,74.93,47.55,23.58,21.97,6.73,4.73.HRMS(m/z):C18H28BrIO2SiNa+[M+Na]+:532.9979,532.9974.
实施例24
TMS乙炔(42uL,0.30mmol),Pd(PPh3)4(11.54mg,0.01mmol)和CuI(3.8mg,0.02mmol)加入到化合物28(50mg,0.10mmol)的THF(2mL)溶液中。随后,Et3N(14uL,0.10mmol)加入,并反应2.5小时。减压旋走低沸点溶剂。所剩物质溶液MeOH(5mL)中,并加入K2CO3(40mg,0.29mmol)。体系搅拌1小时后,饱和的NaHCO3溶液(20mL)猝灭反应。分液后水相用乙酸乙酯(3x30mL)反复萃取。合并有机相,用饱和食盐水(30mL)洗涤,无水Na2SO4干燥。旋干后硅胶柱分离获得无色油状物28-1(36mg,90%)无色油状物.TLC:Rf=0.20(硅胶,3%乙酸乙酯的正己烷溶液).
0℃条件下,咪唑(10mg,0.15mmol)和TBSCl(25mg,0.17mmol)逐次加入到28-1(12mg,0.03mmol)的DCM(3mL)中。反应升至室温,继续反
Callyspongiolide立体异构体的合成及抗癌应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



















动态评分
0.0