

IPC分类号 : C07C49/80,C07C49/84,C07C45/63,C07C205/45,C07C201/12,C07B39/00





专利摘要
本发明属于化工技术领域,具体涉及一种高溴原子利用率的制备α‑单溴代芳香酮的方法。该方法以芳香酮类化合物为底物,在空气氛围下,在H+、NO3‑、I‑存在下,向体系中滴加Br2,在20℃~70℃下反应,得到α‑单溴代芳香酮。本发明采用空气作氧化剂,在H+、NO3‑、I‑存在下,对溴素和芳香酮类化合物进行溴代反应产生的副产物溴化氢进行了原位氧化和选择性溴代反应,以高收率、高选择性、更加环保的优势制得了α‑单溴代芳香酮,适合于大规模工业化生产。
权利要求
1.一种制备α-单溴代芳香酮的方法,其特征在于,以芳香酮类化合物为底物,在空气氛围下,在H
所述芳香酮类化合物、Br
所述芳香酮类化合物为苯乙酮、对硝基苯乙酮、邻硝基苯乙酮、对甲氧基苯乙酮或对氯苯乙酮中的一种;
所述H
所述NO
所述I
2.根据权利要求1所述的制备方法,其特征在于,当芳香酮类化合物在室温下为固体时,先加入有机溶剂将芳香酮类化合物溶解后再进行溴代反应。
3.根据权利要求2所述的制备方法,其特征在于,有机溶剂为二氯甲烷、1,2-二氯乙烷、硝基苯或氯苯中的一种。
说明书
技术领域
本发明属于化工技术领域,具体涉及一种高溴原子利用率的制备α-单溴代芳香酮的方法。
背景技术
α-单溴代芳香酮在是精细化工合成中重要的中间体,用于医药、农药、染料等领域,例如α-溴代对硝基苯乙酮是合成医药氯霉素的原料。
传统上,工业中制备α-单溴代酮通常采用Br2对酮的α-H进行取代。这需要使用与酮相等摩尔量或更多的溴、同时产生等摩尔量的HBr,溴的利用率最多仅达50%。尽管这一方法的原料成本较低,但是产生的大量的废液需进一步处理。为提高上述反应中溴的原子利用率,可采用氧化剂将反应中产生的HBr氧化成Br2、接着对酮进行溴化反应,或以Br
发明内容
为弥补现有技术的不足,本发明目的在于提供一种以Br2为溴源,高溴原子利用率、低成本的对芳香酮溴化制备α-单溴代芳香酮的方法。
本发明的合成方法如式1所示:
本发明采用的技术方案是:以芳香酮类化合物为底物,在H
所述芳香酮类化合物为苯乙酮、对硝基苯乙酮、邻硝基苯乙酮、对甲氧基苯乙酮或对氯苯乙酮中的一种;
所述无机酸为H2SO4、HNO3、H3PO4或HI中的一种;
所述NO3
所述I
优选地,所述方法中芳香酮类化合物、Br2、I
优选地,所述反应温度为初始滴加Br2的温度为20℃~50℃,加完Br2后反应温度控制在30℃~70℃。
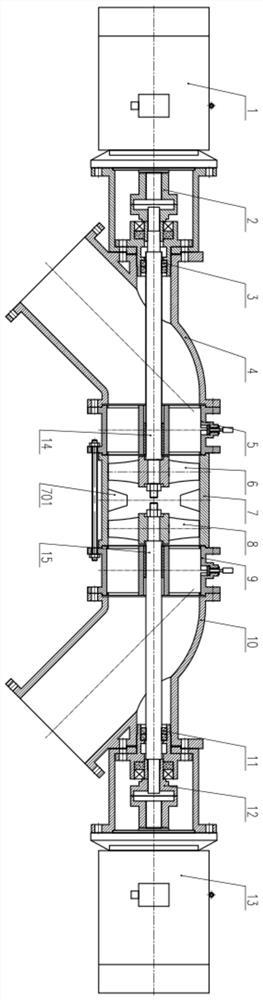
更为具体地,α-单溴代芳香酮的制备方法为:在空气气氛下,将芳香酮类化合物、H
与现有技术相比本发明的有益效果主要体现在:
(1)本发明通过Br2、HNO3形成的氧化循环体系提高了Br2的利用率,达到了95%以上,明显降低了生产中的原料溴的成本;
(2)本发明提高了对α-单取代芳香酮的选择性,得到了高收率的α-单溴取代酮;
(3)本发明不使用过渡金属离子,避免了对芳香环的溴代反应,避免了现有技术中用到的铜催化剂生成溴化亚铜副产物及其后处理问题;
(4)本发明有效缩短了反应时间;
(5)本发明反应后的产物所在的有机相和其它无机物及其副产物所在的水相的分离可通过简单相分离实现;水相可直接循环,进行下一批酮的溴化反应。
具体实施方式
下面通过具体实例详述本发明,但不限制本发明的保护范围。如无特殊说明,本发明所采用的实验方法均为常规方法,所用实验器材、材料、试剂等均可从化学公司购买。
实施例1
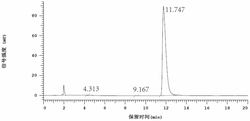
将苯乙酮(7.0mL,60mmol)、H2SO4(15.0mL,6M)、NaNO3(0.76g,8.9mmol)、KI(1.00g,6.0mmol)、氯苯(100mL)加入三口烧瓶中,搅拌,在30℃下滴加Br2(1.56mL,30mmol),约0.5h滴毕。升温至50℃搅拌,反应1h后静置后分层。上层水相转移到反应瓶可循环利用,下层有机相经亚硫酸氢钠溶液洗涤、碳酸氢钠溶液洗涤、干燥、HPLC分析,苯乙酮:α-溴苯乙酮:α,α-二溴苯乙酮之摩尔比为2.5%:96.1%:1.4%。有机相经减压脱溶、重结晶得到α-溴苯乙酮,10.8g,收率91%。熔点47-48℃,文献值47–48℃
实施例2
将苯乙酮(7.0mL,60mmol)、H3PO4(5.0mL,3M)KNO3(0.61g,6.0mmol)、KI(1.00g,6.0mmol)、氯苯(100mL)加入三口烧瓶中,搅拌溶解,在30℃滴加Br2(1.56mL,30mmol),约0.5h滴毕。升温至40℃搅拌,用TLC跟踪反应进程,反应6h静置后分层,下层有机相经亚硫酸氢钠溶液洗涤、碳酸氢钠溶液洗涤、干燥、HPLC分析,苯乙酮:α-溴苯乙酮:α,α-二溴苯乙酮之摩尔比为10.5%:89.3%:0.2%。可通过分离提高一溴苯乙酮的含量,收率85%。
实施例3
将对硝基苯乙酮(10.0g,60.6mmol)、H2SO4(10mL,6M)、HNO3(7.0mL,3M)、KI(0.50g,3.0mmol)氯苯(100mL)加入三口烧瓶中,搅拌溶解,在40℃下滴加Br2(1.60mL,31mmol),约0.5h滴毕,升温至60℃加热搅拌,反应3h后停止反应,静置后分层。上层水相倒入反应瓶可循环利用,下层有机相经亚硫酸氢钠溶液洗涤、碳酸氢钠溶液洗涤、干燥、经HPLC分析,对硝基苯乙酮:α-溴代对硝基苯乙酮:α,α-二溴代对硝基苯乙酮之摩尔比为5:88.2:6.8。有机相经减压脱溶、重结晶得到α-溴代对硝基苯乙酮,11.85g,收率80%。熔点99-100℃,文献值96-97℃
实施例4
将对甲氧基苯乙酮(9.0g,60mmol)、H2SO4(5mL,6M)、HNO3(2.0mL,3M)、NaI(0.80g,6.0mmol)、氯苯(100mL)加入到三口烧瓶中,搅拌,在20℃下滴加Br2(1.55mL,30mmol),约0.5h滴毕,搅拌并缓慢通入空气,TLC监测,反应,3h后停止反应,静置后分出有机相。有机相经亚硫酸氢钠溶液洗涤、碳酸氢钠溶液洗涤、干燥。HPLC分析,对甲氧基苯乙酮:α-溴代对甲氧基苯乙酮:α,α-二溴代甲氧基苯乙酮之摩尔比为1.2:96.2:2.6。有机相经减压脱溶、重结晶得到α-溴代对甲氧基苯乙酮,12.5g,收率91%。
实施例5
向三口烧瓶中加入对甲氧基苯乙酮(9.0g,60mmol)、H3PO4(9.0mL,3M)、HNO3(1.0mL,3M)、NaI(0.80g,6.0mmol)、二氯甲烷(100mL)加入三口瓶中,搅拌,在30℃下滴加Br2(1.55mL,30mmol),约0.5h滴毕,在30℃下继续加热搅拌并缓慢通入空气,TLC监测反应,1.3h后停止反应,静置分出有机相,经亚硫酸氢钠溶液洗涤、碳酸氢钠溶液洗涤、干燥、HPLC分析,对甲氧基苯乙酮:α-溴代对甲氧基苯乙酮:α,α-二溴代对甲氧基苯乙酮之摩尔比为1.2:96.2:2.6。有机相经减压脱溶、重结晶得到α-溴代对甲氧基苯乙酮,12.5g,收率91%。
实施例6
将对氯苯乙酮(7.72g,50mmol)、H2SO4(5mL,6M)、NaI(0.40g,3.0mmol)NaNO3(0.64g,7.5mmol)、氯苯(80mL)加入三口烧瓶,搅拌,在40℃下滴加Br2(1.37mL,26mmol),约0.5h滴毕,之后缓慢通入空气,在60℃下加热搅拌,TLC监测,反应,反应2h后冷至室温,分离有机相,经亚硫酸氢钠溶液洗涤、碳酸氢钠溶液洗涤、干燥、减压脱溶、重结晶得到α-溴代对氯苯乙酮11.0g,收率为93%。熔点96-97℃,文献值95-96℃
实施例7
将对氯苯乙酮(7.72g,50mmol)、磷酸(5mL,6M)、KI(0.83g,5.0mmol)、氯苯(80mL)加入三口烧瓶中,搅拌,在40℃下滴加Br2(1.37mL,27mmol),同时滴加HNO3(5.0mL,3M),约0.5h滴毕,在60℃下加热搅拌,GC监测,反应,反应2h后冷至室温,分离有机相,经亚硫酸氢钠溶液洗涤、碳酸氢钠溶液洗涤、干燥、减压脱溶、重结晶得到α-溴代对氯苯乙酮8.42g,收率为71%。
对比例1
文献
对比例2
文献
对比例3
文献
对比例4
文献
对比例5
文献
对比例6
将对甲氧基苯乙酮9.0g(60mmol)、二氯乙烷(100mL)加入到三口烧瓶中,在40℃下加热搅拌至溶解,滴加Br2(1.55mL,30mmol),约0.5h滴毕;缓慢通入空气,继续搅拌反应4h,取出0.1mL有机相,经二氯乙烷稀释、先后经亚硫酸氢钠溶液洗涤、碳酸氢钠溶液洗涤、干燥处理后进行HPLC分析。对甲氧基苯乙酮:α-溴代对甲氧基苯乙酮:α,α-二溴代对甲氧基苯乙酮之摩尔比为47.1%:52.2%:0.7%。
对比例7
将对甲氧基苯乙酮9.0g(60mmol)、二氯乙烷(100mL)、NaNO3(0.51g,6.0mmol)、H2O(5mL)加入到三口烧瓶中,在40℃下加热搅拌至溶解,滴加Br2(1.55mL,30mmol),约0.5h滴毕;缓慢通入空气,继续搅拌反应4h,分离出的有机相经亚硫酸氢钠溶液洗涤、碳酸氢钠溶液洗涤、干燥处理后进行HPLC分析。对甲氧基苯乙酮:α-溴代对甲氧基苯乙酮:α,α-二溴代对甲氧基苯乙酮之摩尔比为14.6%:81.6%:3.8%。
由上述实施例和对比例可知本发明加入H
参考文献:
1.(a)李景华,牛宗强,王剑,孙健,一种α-溴代芳香酮类化合物的合成方法,CN102503751A。
(b)Jianqiang Wang,Xiaolei Wang,Zong-Qiang Niu,Jian Wang,Man Zhang,andJing-Hua Li,Copper nitrate–catalyzedα-bromination of aryl ketones withhydrobromic acid,Synth.Commun.,2016,46,165–168.
2.张书文李志云,一种过氧化氢氧化溴化法合成α-溴代酮类化合物的方法,CN101928208A。
3.Vasudha H.Tillu,Popat D.Shinde,Ashu tosh V.Bedekar,and RadhikaD.Wakharkar,Studies on Bromination of Active Methylene by a Mixture ofHydrobromic Acid and Hydrogen Peroxide(or TBHP),Synth.Commun.,2003,33,1399–1403.
4.A.Podgorsek,S.Stavber,M.Zupan,J.Iskra,Bromination of ketones withH2O2–HBr“on water”,Green Chem.,2007,9,1212–1218.
5.Archana K.Ghorpade,Sameerana N.Huddar,Krishnacharya G.Akamanchi,AqHBr–NaNO2–KI/air:a new catalytic system for α-monobromination of ketones,Tetrahedron Letters,2016,57,4918–4921.
6.韩冰冰,牛青龙,吴新虎,何在明,吴成凤,郑祖彪,一种由酮类化合物选择性溴化制备ɑ-单溴代酮和ɑ,ɑ-二溴代酮类化合物的方法,CN104119211A。
以上所述,仅为本发明创造较佳的具体实施方式,但本发明创造的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明创造披露的技术范围内,根据本发明创造的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明创造的保护范围之内。
一种制备α-单溴代芳香酮的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。


















动态评分
0.0