





专利摘要
本发明涉及孔材料领域,公开了一种磷酸铌孔材料及其制备方法和用途。所述磷酸铌孔材料包括铌元素、磷元素和氧元素,其孔容在0.3cm3/g以上、平均孔径在9nm以上、氮气等温吸脱附测试下具有3‑60nm的孔径分布、5.6‑15.8nm范围的孔径占总孔径分布量的比例≥80%。制备磷酸铌孔材料的方法包括:在溶剂的存在下,将可溶性铌盐、磷源和模板剂混合后进行超声处理,再将超声处理后得到的固相进行焙烧。本发明还公开了由上述方法制得的磷酸铌孔材料。此外,本发明还公开了所述磷酸铌孔材料在催化分子内脱水反应中的用途。本发明通过操作简易的方法得到了孔径较大且孔径分布较窄的磷酸铌孔材料,其可以作为固体酸催化剂使用。
权利要求
1.一种磷酸铌孔材料,其特征在于,该磷酸铌孔材料包括:铌元素、磷元素和氧元素,其中,所述磷酸铌孔材料的孔容在0.3cm3/g以上、平均孔径在9nm以上、氮气等温吸脱附测试下具有3-60nm的孔径分布、5.6-15.8nm范围的孔径占总孔径分布量的比例≥80%。
2.根据权利要求1所述的磷酸铌孔材料,其中,所述磷酸铌孔材料的孔容为0.3-0.5cm3/g、平均孔径为9-10.6nm、氮气等温吸脱附测试下5.6-15.8nm范围的孔径占总孔径分布量的比例80-95%。
3.根据权利要求1所述的磷酸铌孔材料,其中,所述磷酸铌孔材料的比表面积为140-300m2/g。
4.根据权利要求1-3中任意一项所述的磷酸铌孔材料,其中,铌元素与磷元素的摩尔比为1:0.4-2.6。
5.一种制备磷酸铌孔材料的方法,该方法包括:在溶剂的存在下,将可溶性铌盐、磷源和模板剂混合后进行超声处理,再将超声处理后得到的固相进行焙烧。
6.根据权利要求5所述的方法,其中,相对于每摩尔的可溶性铌盐,所述磷源的用量为0.4-2.6mol,所述模板剂的用量为3-120g,所述可溶性铌盐以铌元素计,所述磷源以磷元素计。
7.根据权利要求5或6所述的方法,其中,所述可溶性铌盐为五氯化铌、草酸铌、苹果酸铌、酒石酸铌和柠檬酸铌中的至少一种;和/或
所述溶剂为水、甲醇和乙醇中的至少一种。
8.根据权利要求5或6所述的方法,其中,所述可溶性铌盐为柠檬酸铌且所述柠檬酸铌通过以下方法制得:
将铌源与氢氟酸接触反应,直至反应体系变得澄清;将得到的澄清液与碱性物质接触,再将接触后得到的固相在溶剂的存在下与柠檬酸混合,直至混合体系变得澄清,其中,相对于每摩尔的铌元素,柠檬酸的用量为1.7-10mol。
9.根据权利要求5所述的方法,其中,所述磷源为磷酸盐、磷酸二氢盐和磷酸氢盐中的至少一种。
10.根据权利要求5所述的方法,其中,所述模板剂为阳离子表面活性剂和/或非离子表面活性剂;或者
所述模板剂为碳氢表面活性剂和/或氟碳表面活性剂。
11.根据权利要求5所述的方法,其中,混合可溶性铌盐、磷源和模板剂的方法为:将可溶性铌盐的溶液滴加至含有磷源和模板剂的混合溶液中,其中,含有磷源和模板剂的混合溶液的pH值为1-10。
12.根据权利要求5-11中任意一项所述的方法,其中,所述超声处理的条件包括:频率为25-130KHz,功率密度为80-1200w/cm2,温度为20-100℃,pH值为1-7,时间为0.5-4h。
13.根据权利要求5-11中任意一项所述的方法,其中,所述超声处理的条件包括:频率为25-45KHz,功率密度为100-300w/cm2,温度为20-50℃,pH值为1-7,时间为1.5-3h。
14.根据权利要求5-11中任意一项所述的方法,其中,所述焙烧的条件包括:温度为360-550℃,时间为3-5h。
15.由权利要求5-14中任意一项所述的方法制得的磷酸铌孔材料。
16.权利要求1-4和15中任意一项所述的磷酸铌孔材料在催化分子内脱水反应中的用途。
说明书
技术领域
本发明涉及孔材料(分子筛)领域,具体地,涉及一种磷酸铌孔材料及其制备方法和用途。
背景技术
铌及其化合物是一种重要的功能材料,广泛应用于冶金、超导材料等领域。近年来,铌化合物,尤其是铌酸,作为催化组分在科研和工业中的应用越来越多。然而铌酸极低的比表面积和非孔结构,大大限制了其催化性能。而磷酸铌是一种优异的固体酸材料,而具有孔道结构的大比表面积的磷酸铌材料则显示出重要的研究价值和应用前景。
CN102962085A和CN102951683A公开了一种含有介孔结构的磷酸铌分子筛。其特征在于其材料具有介孔结构,BET比表面积为160-290m2/g,孔容为0.16-0.29cm3/g。
水热合成反应是介孔分子筛制备的常规方法,广泛用于各种分子筛包括介孔分子筛材料的合成。然而,传统的水热合成法需要相对较高的温度和相对较长的晶化时间,耗能费时,而且,传统方法难以制得孔径较大且孔径分布较窄的磷酸铌孔材料。因此,研究操作简易的工艺来制备具有较大孔径且孔径分布较窄的磷酸铌孔材料具有重要的意义。
发明内容
本发明的目的是克服现有工艺能耗高操作复杂的缺陷,提供一种孔径较大且孔径分布较窄的磷酸铌孔材料及其简易制备方法和在催化分子内脱水反应中的用途。
为了实现上述目的,第一方面,本发明提供了一种磷酸铌孔材料,该磷酸铌孔材料包括:铌元素、磷元素和氧元素,其中,所述磷酸铌孔材料的孔容在0.3cm3/g以上、平均孔径在9nm以上、氮气等温吸脱附测试下具有3-60nm的孔径分布、5.6-15.8nm范围的孔径占总孔径分布量的比例≥80%。
第二方面,本发明提供了一种制备磷酸铌孔材料的方法,该方法包括:在溶剂的存在下,将可溶性铌盐、磷源和模板剂混合后进行超声处理,再将超声处理后得到的固相进行焙烧。
第三方面,本发明提供了由第二方面所述的方法制得的磷酸铌孔材料。
第四方面,本发明提供了第一方面和/或第二方面所述的磷酸铌孔材料在催化分子内脱水反应中的用途。
本发明通过操作简易的方法得到了孔径较大且孔径分布较窄的磷酸铌孔材料,该磷酸铌孔材料可以作为固体酸催化剂使用,例如,用于有效地催化分子内脱水反应。
本发明的其它特征和优点将在随后的具体实施方式部分予以详细说明。
附图说明
附图是用来提供对本发明的进一步理解,并且构成说明书的一部分,与下面的具体实施方式一起用于解释本发明,但并不构成对本发明的限制。在附图中:
图1是根据本发明的一种实施方式的磷酸铌孔材料的氮气吸脱附曲线图;
图2是根据本发明的一种实施方式的磷酸铌孔材料的透射电子显微镜(TEM)照片;
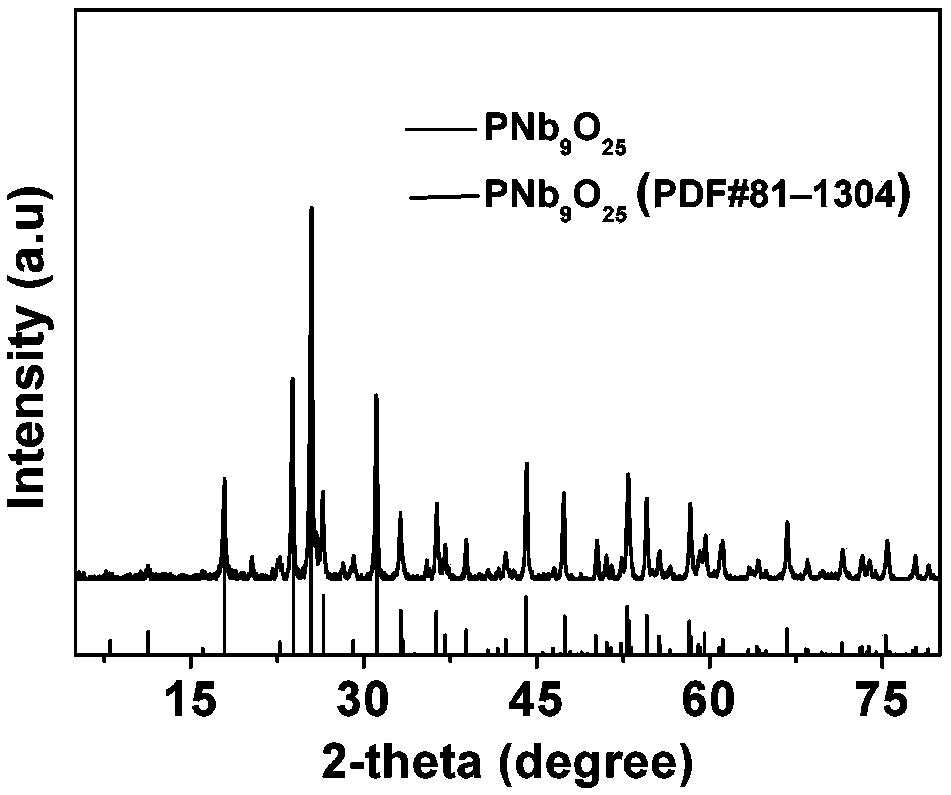
图3是根据本发明的一种实施方式的磷酸铌孔材料的X-射线衍射(XRD)谱图。
具体实施方式
以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
本发明提供的磷酸铌孔材料包括:铌元素、磷元素和氧元素,其中,所述磷酸铌孔材料的孔容在0.3cm3/g以上、平均孔径在9nm以上、氮气等温吸脱附测试下具有3-60nm的孔径分布、5.6-15.8nm范围的孔径占总孔径分布量的比例≥80%。
磷酸铌孔材料的孔容指孔材料内的孔体积,平均孔径指平均孔内直径,这为本领域技术人员所熟知,此处不赘述。
优选地,所述磷酸铌孔材料的孔容为0.3-0.5cm3/g。
优选地,所述磷酸铌孔材料的平均孔径为9-10.6nm。
优选地,所述磷酸铌孔材料在氮气等温吸脱附测试下5.6-15.8nm范围的孔径占总孔径分布量的比例80-95%。本发明中5.6-15.8nm范围的孔径占总孔径分布量的比例可以按如下公式计算:5.6-15.8nm范围的孔径的数量/3-60nm范围的孔径的数量×100%。
优选地,所述磷酸铌孔材料的比表面积为140-300m2/g。其中,磷酸铌孔材料的比表面积是指BET总比表面积,可以按照ASTM D4222-98标准方法测得。
根据本发明的优选实施方式,磷酸铌孔材料中铌元素与磷元素的摩尔比为1:0.4-2.6,进一步优选地,磷酸铌孔材料中铌元素与磷元素的摩尔比为1:0.45-1.5(如1:0.45、1:0.6、1:0.8、1:1、1:1.2、1:1.3、1:1.4、1:1.5或前述数值之间的任意值)。
本发明对前述磷酸铌孔材料的制备方法无特殊要求,只要能够制备得到具有上述结构的磷酸铌孔材料即可,根据本发明的一种优选的实施方式,本发明采用超声处理的方法制备前述磷酸铌孔材料。
因此,本发明提供的制备磷酸铌孔材料的方法包括:在溶剂的存在下,将可溶性铌盐、磷源和模板剂混合后进行超声处理,再将超声处理后得到的固相进行焙烧。
本发明中,在未作相反说明的情况下,本发明中的可溶性铌盐的用量以铌元素计,磷源的用量以磷元素计。
根据本发明的优选实施方式,所述可溶性铌盐与磷源的摩尔比为1:0.4-2.6,更优选为1:0.45-1.5(如1:0.45、1:0.6、1:0.8、1:1、1:1.2、1:1.3、1:1.4、1:1.5或前述数值之间的任意值)。
根据本发明的优选实施方式,相对于每摩尔的可溶性铌盐(以铌元素计),所述模板剂的用量为3-120g(如4g、15g、25g、35g、45g、55g、65g、75g、85g、95g、105g、115g或前述数值之间的任意值),更优选为35-75g。
根据本发明的优选实施方式,相对于每摩尔的可溶性铌盐,溶剂的用量为2-20L,更优选为5-10L。
根据本发明,所述可溶性铌盐可以为本领域常见的各种能够提供铌元素的可溶性物质,可以为有机可溶性铌盐,也可以为无机可溶性铌盐。优选情况下,所述可溶性铌盐为五氯化铌、草酸铌(例如,参见CN1935772A或CN1911892A)、苹果酸铌、酒石酸铌(例如,参见CN102951683A)和柠檬酸铌中的至少一种。
根据本发明的一种优选实施方式,所述可溶性铌盐为(水溶性)柠檬酸铌且所述柠檬酸铌通过以下方法制得(换言之,所述方法还包括通过以下方法制备柠檬酸铌的步骤):
将铌源与氢氟酸接触反应,直至反应体系变得澄清;将得到的澄清液与碱性物质接触,再将接触后得到的固相在溶剂的存在下与柠檬酸混合,直至混合体系变得澄清,其中,相对于每摩尔的铌元素,柠檬酸的用量为1.7-10mol(1.7mol、1.8mol、1.9mol、2mol、2.5mol、3mol、3.5mol、4mol、4.5mol、5mol、5.5mol、6mol、6.5mol、7mol、7.5mol、8mol、8.5mol、9mol、9.5mol、10mol或前述数值之间的任意值,优选为1.9-5mol)。
上述柠檬酸铌的制备方法中,对氢氟酸的用量没有特别的限定,只要能够充分溶解铌源即可。相对于每摩尔的铌元素,所述氢氟酸以氟化氢计的用量优选为5-15mol。氢氟酸中氟化氢的含量通常为2-15mol/L。
上述柠檬酸铌的制备方法中,对碱性物质的用量没有特别的限定,只要能够与澄清液接触后形成沉淀即可。相对于每摩尔的铌元素,碱性物质的用量优选为5-50mol,更优选为9-25mol/L。
优选地,混合体系中铌元素的浓度为0.01-3mol/L(如0.01mol/L、0.05mol/L、0.09mol/L、0.1mol/L、0.15mol/L、0.2mol/L、0.25mol/L、0.3mol/L、0.5mol/L、1mol/L、2mol/L、3mol/L或前述数值之间的任意值),更优选为0.05-1.5mol/L。
上述柠檬酸铌的制备方法中,所述铌源可以为本领域常见的各种铌源(即能够提供铌元素的物质)。优选情况下,所述铌源为铌的氧化物、铌酸盐、氟铌酸盐和铌酸中的至少一种,更优选为五氧化二铌、铌酸钾、铌酸钠、铌酸钙、氟铌酸钾(即七氟铌酸钾)和氟铌酸钠中的至少一种。“铌酸”指水合的氧化铌,制备方法可以参见文献“J.D.A.Goncalves,A.L.D.Ramos,L.L.Rocha,A.K.Domingos,R.S.Monteiro,J.S.Peres,N.C.Furtado,C.A.Taft,D.A.G.Aranda,Niobium oxide solid catalyst:esterification of fatty acids and modeling for biodiesel product.J.Phy.Organic.Chem.,2011,24(1):54-64.”。
上述柠檬酸铌的制备方法中,所述碱性物质可以为无机碱,也可以为有机碱,例如,在25℃下1mol/L水溶液的pH值大于11的物质。优选地,所述碱性物质为氨水。
上述柠檬酸铌的制备方法中,铌源与氢氟酸接触反应的条件可以包括:温度为50-80℃,时间为1-20h。
上述柠檬酸铌的制备方法中,澄清液与碱性物质接触的条件可以包括:温度为25-80℃,时间为5-60min。
上述柠檬酸铌的制备方法中,固相与柠檬酸混合的条件可以包括:温度为25-100℃,时间为5-720min(优选30-60min)。一般地,固相经洗涤后再与柠檬酸混合。可以使用去离子水或稀氨水进行洗涤。
上述柠檬酸铌的制备方法中,所述方法还可以包括:将(与柠檬酸)混合后的产物与铵离子源接触,再搅拌直至混合体系变得澄清。通过使用铵离子源,能够进一步促进混合体系变得澄清,并使得到的柠檬酸铌更适于制备性能较佳的含铌催化剂。相对于每摩尔的铌元素,所述铵离子源以铵离子计的用量为0.01-30mol(0.01mol、0.05mol、0.08mol、0.1mol、0.2mol、0.5mol、1mol、2mol、5mol、10mol、15mol、20mol、25mol、30mol或前述数值之间的任意值,优选为0.01-0.1mol)。所述铵离子源可以为各种常规的能够提供铵离子的物质,优选地,所述铵离子源为氨水和/或铵盐,更优选为氨水和/或硝酸铵。
上述柠檬酸铌的制备方法中,对所述溶剂没有特别的要求,可以为常规的溶剂,如水。
上述柠檬酸铌的制备方法中,为了获得固体形式的柠檬酸铌,本发明的方法还可以包括:使混合体系变澄清后的产物结晶。对结晶的具体方法没有特别的要求,例如,可以为冷却结晶或蒸发结晶。本领域技术人员能够对结晶的条件进行选择,在此不再赘述。
为了获得更易于储存的柠檬酸铌,本发明的方法还可以包括将结晶后的产物进行干燥的步骤。干燥的条件可以为常规的条件,如50-150℃下干燥10-20h。
根据本发明,所述磷源可以为各种能够提供磷元素的可溶性物质,例如,磷酸盐、磷酸二氢盐和磷酸氢盐中的至少一种。优选情况下,所述磷源为磷酸二氢钠、磷酸二氢钾、磷酸二氢铵、磷酸钠、磷酸钾、磷酸铵、磷酸氢二铵、磷酸氢二钠和磷酸氢二钾中的至少一种。
根据本发明,所述模板剂可以为本领域常规使用的各种模板剂,如表面活性剂。优选地,所述模板剂为阳离子表面活性剂(如十六烷基三甲基溴化铵,CTAB)、非离子表面活性剂(如P123)、和氟碳表面活性剂(如FS-3100)中的至少一种。
根据本发明,只要在溶剂的存在下将可溶性铌盐、磷源和模板剂的混合物进行超声处理即可实现本发明的目的,对可溶性铌盐、磷源和模板剂的混合方式没有特别的要求,但优选情况下,将混合可溶性铌盐、磷源和模板剂的方法为:将可溶性铌盐的溶液滴加至含有磷源和模板剂的混合溶液中,其中,含有磷源和模板剂的混合溶液的pH值为1-10(优选为2-7)。
在制备磷酸铌孔材料的方法中,所述溶剂可以为水、甲醇和乙醇中的至少一种。本发明中制备磷酸铌孔材料时使用的溶剂与制备柠檬酸铌时使用的溶剂可以相同或不同。
根据本发明的优选实施方式,所述超声处理的条件包括:频率为25-130KHz,更优选为25-45KHz。
根据本发明的优选实施方式,所述超声处理的条件还包括:功率密度为80-1200w/cm2,更优选为100-300w/cm2。
根据本发明的优选实施方式,所述超声处理的条件还包括:温度为20-100℃,更优选为20-50℃。
根据本发明的优选实施方式,所述超声处理的条件还包括:pH值为1-7。
根据本发明的优选实施方式,所述超声处理的条件还包括:时间为0.5-4h,更优选为1.5-3h。
根据本发明,可以采用本领域常规的条件进行焙烧。优选地,所述焙烧的条件包括:温度为360-550℃。优选地,所述焙烧的条件还包括:时间为3-5h。焙烧的气氛包括空气气氛。更优选地,采用先在惰性气氛中使待焙烧的物料升温至焙烧温度,然后转至空气气氛中进行焙烧,因此,所述焙烧的条件包括:首先在360-550℃于惰性气氛中焙烧0.5-2h,然后在360-550℃于空气气氛中焙烧0.5-5h。
本领域技术人员所熟知的是,焙烧前,可以对超声处理后得到的固相进行洗涤和干燥,在此不再赘述。
此外,本发明还提供了由上述方法制得的磷酸铌孔材料。如前所述,所得磷酸铌孔材料的孔容在0.3cm3/g以上、平均孔径在9nm以上、氮气等温吸脱附测试下具有3-60nm的孔径分布、5.6-15.8nm范围的孔径占总孔径分布量的比例≥80%,不再逐一赘述。
另外,本发明还提供了磷酸铌孔材料在催化分子内脱水反应(如催化葡萄糖转化为5-羟甲基糠醛)中的用途。
以下将通过实施例对本发明进行详细描述。以下实施例中,样品的孔容、孔径分布、比表面积在Micromeritics公司ASAP2405静态氮吸附仪上测定;电感耦合等离子体分析在珀金埃尔默公司的Optima 8300型电感耦合等离子光谱发生仪上进行;总碳分析在岛津TOC-L型总碳分析仪上进行。
制备例1
该制备例用来制备实施例中使用的可溶性铌盐。
称取4g五氧化二铌,加入40mL氢氟酸(浓度为7mol/L)中,加热搅拌直至溶解变澄清(50℃,3h);将溶液加入150mL氨水(浓度为2mol/L)得到白色沉淀(30℃,1h);将白色沉淀洗涤过滤,然后加入120mL浓度为0.5mol/L的柠檬酸水溶液中,25℃下搅拌0.5h得到澄清溶液,即为柠檬酸铌溶液,通过电感耦合等离子体(ICP)和总碳(TOC)分析可知该溶液中柠檬酸和铌的摩尔比为1.98。将得到的柠檬酸铌溶液于60℃下蒸发12h,得到透明果冻状固体(柠檬酸铌)。
称取4g氟铌酸钾,加入40mL氢氟酸(浓度为3.5mol/L)中,加热搅拌直至溶解变澄清(80℃,5h);将溶液加入150mL氨水(浓度为1mol/L)得到白色沉淀(80℃,5min);将白色沉淀洗涤过滤,然后加入130mL浓度为0.5mol/L的柠檬酸水溶液和10mL氨水(浓度为0.1mol/L),60℃下搅拌0.5h得到澄清溶液,即为柠檬酸铌溶液-1。通过ICP和TOC分析可知,溶液中铵离子、柠檬酸和铌的摩尔比为0.08:4.89:1。
称取4g铌酸钠,加入40mL氢氟酸(浓度为9mol/L)中,加热搅拌直至溶解变澄清(80℃,3h);将溶液加入300mL氨水(浓度为2mol/L)得到白色沉淀(30℃,1h);将白色沉淀洗涤过滤,然后加入150mL浓度为0.5mol/L的柠檬酸水溶液和10mL氨水(浓度为0.1mol/L),80℃下搅拌1h得到澄清溶液,即为柠檬酸铌溶液-2。通过ICP和TOC分析可知,溶液中铵离子、柠檬酸和铌的摩尔比为0.03:3.12:1。
按照CN1935772A实施例1的方法制备草酸铌。
实施例1
称取1.2g十六烷基三甲基溴化铵(CTAB)和3.64g磷酸氢二钠溶于70mL去离子水,充分搅拌溶解,用盐酸调节pH值到2,得到溶液A。称取柠檬酸铌8g(含铌1.84g)溶于42.6mL去离子水中,充分搅拌溶解,得溶液B。将B溶液逐滴加入A溶液中。上述混合溶液于20℃下超声处理(频率为40KHz,功率密度为300w/cm2)2h,之后过滤并洗涤,所得固体在100℃下干燥10h,然后在550℃下焙烧5h,得到磷酸铌孔材料样品,氮气等温吸脱附测试下显示样品具有孔结构(氮气吸脱附曲线图见图1)且具有3-60nm的孔径分布,其它参数测定结果见表1。
将样品置于TEM下进行观察,从TEM照片(图2)可以看出样品具有蠕虫状孔结构,孔径在9nm左右。
对制得的样品在Siemens D5005型X-射线衍射仪上进行XRD分析(下同),采用Cu靶Kα(λ=0.154056nm)源,测试电压为40kV,测试电流为40mA,扫描范围10-80°,扫描速度6°/min,根据XRD谱图(见图3)判断所得样品具有长程无序的介孔结构。
对比例1
本对比例用来说明常规水热法制备磷酸铌孔材料的方法。
称取1.2g十六烷基三甲基溴化铵(CTAB)和3.64g磷酸氢二钠溶于70mL去离子水,充分搅拌溶解,用盐酸调节pH值到2,得到溶液A。称取柠檬酸铌8g(含铌1.84g)溶于42.6mL去离子水中,充分搅拌溶解,得溶液B。将B溶液逐滴加入A溶液中。上述混合溶液在90℃下搅拌老化24h,然后转移到带聚四氟乙烯内衬的不锈钢压力容弹内,在170℃下晶化72h。之后过滤并洗涤,所得固体在100℃下干燥10h,然后在550℃下焙烧5h,得到磷酸铌孔材料样品,氮气等温吸脱附测试下显示样品具有3-60nm的孔径分布,其它参数测定结果见表1。从氮气等温吸脱附测试和TEM观察结果可以看出,得到的样品具有蠕虫状孔结构,孔径在3nm左右;从XRD谱图可以看出样品具有长程无序的介孔结构。
实施例2
称取1.2g十六烷基三甲基溴化铵(CTAB)和3.64g磷酸氢二钠溶于70mL去离子水,充分搅拌溶解,用盐酸调节pH值到2,得到溶液A。称取柠檬酸铌8g(含铌1.84g)溶于42.6mL去离子水中,充分搅拌溶解,得溶液B。将B溶液逐滴加入A溶液中。上述混合溶液于20℃下超声处理(频率为40KHz,功率密度为100w/cm2)2h,之后过滤并洗涤,所得固体在100℃下干燥10h,然后在氮气气氛下以5℃/min的速率升温至550℃,切换至空气气氛中焙烧5h,得到磷酸铌孔材料样品,氮气等温吸脱附测试下显示样品具有3-60nm的孔径分布,其它参数测定结果见表1。
从氮气等温吸脱附测试和TEM观察结果可以看出,得到的样品具有蠕虫状孔结构,孔径在9nm左右;从XRD谱图可以看出样品具有长程无序的介孔结构。
实施例3
称取1.2g十六烷基三甲基溴化铵(CTAB)和1.82g磷酸氢二钠溶于70mL去离子水,充分搅拌溶解,用盐酸调节pH值到5,得到溶液A。称取五氯化铌5.3g溶于42.6mL去离子水中,充分搅拌溶解,得溶液B。将B溶液逐滴加入A溶液中。上述混合溶液于30℃下超声处理(频率为25KHz,功率密度为300w/cm2)3h,之后过滤并洗涤,所得固体在100℃下干燥10h,然后在550℃下焙烧5h,得到磷酸铌孔材料样品,氮气等温吸脱附测试下显示样品具有3-60nm的孔径分布,其它参数测定结果见表1。
从氮气等温吸脱附测试和TEM观察结果可以看出,得到的样品具有蠕虫状孔结构,孔径在9nm左右;从XRD谱图可以看出样品具有长程无序的介孔结构。
实施例4
称取3g十六烷基三甲基溴化铵(CTAB)和3.64g磷酸氢二钠溶于70mL去离子水,充分搅拌溶解,用盐酸调节pH值到5,得到溶液A。称取草酸铌23g(含铌3.96g)溶于100mL去离子水中,充分搅拌溶解,得溶液B。将B溶液逐滴加入A溶液中。上述混合溶液于50℃下超声处理(频率为45KHz,功率密度为200w/cm2)1.5h,之后过滤并洗涤,所得固体在100℃下干燥10h,然后在550℃下焙烧5h,得到磷酸铌孔材料样品,氮气等温吸脱附测试下显示样品具有3-60nm的孔径分布,其它参数测定结果见表1。
从氮气等温吸脱附测试和TEM观察结果可以看出,得到的样品具有蠕虫状孔结构,孔径在9nm左右;从XRD谱图可以看出样品具有长程无序的介孔结构。
实施例5
按照实施例1的方法制备磷酸铌孔材料,不同的是,用“柠檬酸铌溶液-1”替代“柠檬酸铌”,柠檬酸铌溶液-1的用量使得铌的量为1.84g,氮气等温吸脱附测试下显示样品具有3-60nm的孔径分布,其它参数测定结果见表1。从氮气等温吸脱附测试和TEM观察结果可以看出,得到的样品具有蠕虫状孔结构,孔径在9nm左右;从XRD谱图可以看出样品具有长程无序的介孔结构。
实施例6
按照实施例1的方法制备磷酸铌孔材料,不同的是,用“柠檬酸铌溶液-2”替代“柠檬酸铌”,柠檬酸铌溶液-2的用量使得铌的量为1.84g,氮气等温吸脱附测试下显示样品具有3-60nm的孔径分布,其它参数测定结果见表1。从氮气等温吸脱附测试和TEM观察结果可以看出,得到的样品具有蠕虫状孔结构,孔径在9nm左右;从XRD谱图可以看出样品具有长程无序的介孔结构。
实施例7
按照实施例1的方法制备磷酸铌孔材料,不同的是,用“P123(购自北京伊诺凯科技有限公司)”代替“十六烷基三甲基溴化铵”,氮气等温吸脱附测试下显示样品具有3-60nm的孔径分布,其它参数测定结果见表1。
从氮气等温吸脱附测试和TEM观察结果可以看出,得到的样品具有蠕虫状孔结构,孔径在12nm左右;从XRD谱图可以看出样品具有长程无序的介孔结构。
实施例8
按照实施例1的方法制备磷酸铌孔材料,不同的是,用“FS-3100(购自北京科普佳实验仪器有限公司)”代替“十六烷基三甲基溴化铵”,氮气等温吸脱附测试下显示样品具有3-60nm的孔径分布,其它参数测定结果见表1。从氮气等温吸脱附测试和TEM观察结果可以看出,得到的样品具有蠕虫状孔结构,孔径在11nm左右;从XRD谱图可以看出样品具有长程无序的介孔结构。
表1
测试例1
在间歇反应釜中加入10ml体积比为7:3的甲基异丁基甲酮和氯化钾的饱和水溶液的混合液,然后加入0.5g葡萄糖,分别加入0.1g上述实施例或对比例制得的样品作为固体酸催化剂,充入氮气,加热至160℃,恒温搅拌,反应3小时后将反应体系冷却至室温(25℃),离心分离出催化剂。反应液用高效液相色谱进行分析,计算5-羟甲基糠醛(5-HMF)的收率等。结果如表2所示,其中,转化率=(反应前葡萄糖的摩尔量-反应后葡萄糖的摩尔量)/反应前葡萄糖的摩尔量×100%;5-HMF选择性=生成的5-HMF的摩尔量/(反应前葡萄糖的摩尔量-反应后葡萄糖的摩尔量)×100%;5-HMF收率=转化率×5-HMF选择性。
高效液相色谱分析的条件如下:
反应液使用Agilent 1200型的HPLC分析,色谱柱为XDB-C18色谱柱(4.5μm,250mm,Eclipse USA),色谱柱恒温在35℃。液相色谱装配一个Agilent G1329A型自动进样器,用来增加进样的可重复性。使用AgilentG1314B型紫外检测器(VWD)来检测反应生成的5-HMF产物,紫外波长为254nm,流动相是甲醇和纯水的混合液,体积比为20:80,流速为0.6mL·min-1。采用Agilent 1200型HPLC色谱配置Agilent G1362A型示差折光检测器(RID)和Bio-Rad Aminex HPX-87H糖柱来检测反应后剩余的葡萄糖,色谱柱恒温在80℃,流动相为纯水,流速为0.8mL·min-1。
表2
从以上实施例可以看出,本发明通过操作简易的方法得到了孔径较大且孔径分布较窄的磷酸铌孔材料,该磷酸铌孔材料可以用于有效地催化葡萄糖转化为5-羟甲基糠醛,5-HMF选择性和收率较高。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
一种磷酸铌孔材料及其制备方法和用途专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。














动态评分
0.0