

IPC分类号 : C07C281/16,A61K31/155,A61P31/04,A01N47/44,A01P1/00,C08G73/00,A61K31/785






专利摘要
本发明涉及一种依据如下反应方案由胍、氨基胍或二氨基胍G与一种或更多种苯甲基或烯丙基衍生物BA制备缩聚产物的方法:其中X各自独立地代表离去基团;R1各自独立地代表具有至少一个芳香环的芳香族环体系,所述芳香环任选地包含一个或更多个选自O、N和S的杂原子且任选地被一个或两个乙烯基取代,‑CH2‑X基团被结合至所述乙烯基,或R1各自独立地代表乙烯基;Gua代表胍二基、氨基胍二基或二氨基胍二基;Y代表H‑Gua,并且Z代表H;或Y和Z一起代表化学键,以便得到环状结构;其中使至少一种苯甲基衍生物或烯丙基衍生物BA与过量的胍、氨基胍或二氨基胍G在消除HX的情况下发生缩聚反应。
权利要求
1.一种依据如下反应方案由胍、氨基胍或二氨基胍G与一种或更多种苯甲基或烯丙基衍生物BA制备缩聚产物的方法:
其中
X各自独立地代表离去基团;
R1各自独立地代表具有至少一个芳香环的芳香族环体系,所述芳香环任选地包含一个或更多个选自O、N和S的杂原子且任选地被一个或两个乙烯基取代,-CH2-X基团被结合至所述乙烯基,或R1各自独立地代表乙烯基;
Gua代表胍二基、氨基胍二基或二氨基胍二基;
Y代表H-Gua,并且
Z代表H;或
Y和Z一起代表化学键,以便得到环状结构;
其中使至少一种苯甲基或烯丙基衍生物BA与过量的胍、氨基胍或二氨基胍G在消除HX的情况下发生缩聚反应,以便得到对应于如下化学式(I)、(II)或(III):
或具有通过在消除相应的胍后的环化得到的环状结构的聚胍,或所述聚胍的盐。
2.根据权利要求1所述的方法,其特征在于,R1选自任选地被取代的苯、二乙烯基苯、呋喃、吡咯、噻吩、吡啶、联苯、芴和乙烯的二价残基。
3.根据权利要求2所述的方法,其特征在于,R1选自苯、二乙烯基苯、吡啶、联苯和乙烯的二价残基。
4.根据权利要求1至3中任一项所述的方法,其特征在于,所述离去基团选自氯、溴、碘、甲磺酸酯、三氟甲磺酸酯或甲苯磺酸酯。
5.根据权利要求1至4中任一项所述的方法,其特征在于,通过将反应物在其熔融温度以上的温度加热来使所述至少一种苯甲基或烯丙基衍生物BA与所述胍、氨基胍或二氨基胍G反应。
6.根据权利要求1至5中任一项所述的方法,其特征在于,所述反应被进行持续至少2小时,优选地至少3小时。
7.聚胍,所述聚胍对应于下列化学式(I):
其中R1代表具有至少一个芳香环的芳香族环体系,所述芳香环任选地包含一个或更多个选自O、N和S的杂原子且任选地被一个或两个乙烯基取代,-CH2-X基团被结合至所述乙烯基,或R1代表乙烯基,
或具有通过在消除胍后的环化得到的环状结构。
8.聚胍,所述聚胍对应于下列化学式(II):
其中R1代表具有至少一个芳香环的芳香族环体系,所述芳香环任选地包含一个或更多个选自O、N和S的杂原子且任选地被一个或两个乙烯基取代,-CH2-X基团被结合至所述乙烯基,或R1代表乙烯基,
或具有通过在消除氨基胍后的环化得到的环状结构。
9.聚胍,所述聚胍对应于下列化学式(III):
其中R1代表具有至少一个芳香环的芳香族环体系,所述芳香环任选地包含一个或更多个选自O、N和S的杂原子且任选地被一个或两个乙烯基取代,-CH2-X基团被结合至所述乙烯基,或R1代表乙烯基,
或具有通过在消除二氨基胍后的环化得到的环状结构。
10.根据权利要求7至9中任一项所述的聚胍,其特征在于,R1选自任选地被取代的苯、二乙烯基苯、呋喃、吡咯、噻吩、吡啶、联苯、芴和乙烯的二价残基。
11.根据权利要求10所述的聚胍,其特征在于,R1选自苯、二乙烯基苯、吡啶、联苯和乙烯的二价残基。
12.根据权利要求7至11中任一项所述的聚胍用作抗感染药的用途。
13.用于根据权利要求12所述的用途的聚胍,其特征在于,所述聚胍用于对抗人类或动物患者中的细菌、病毒或真菌感染。
14.用于根据权利要求13所述的用途的聚胍,其特征在于,所述聚胍用于局部施用或全身施用。
15.用于根据权利要求14所述的用途的聚胍,其特征在于,所述聚胍以药品或药物组合物的形式施用。
16.根据权利要求7至11中任一项所述的聚胍用作体外抗微生物剂的用途。
17.根据权利要求16所述的用途,其特征在于,所述聚胍用作抗微生物涂料、涂层、箔或膜的活性组分。
18.用于对抗人类或动物患者中的细菌感染的药品或药物组合物,包含根据权利要求7至11中任一项所述的聚胍作为抗感染药。
19.根据权利要求18所述的药品或药物组合物,其特征在于,其还包含至少一种药学上可接受的载体或赋形剂,以及任选地一种或更多种佐剂和/或一种或更多种另外的活性成分。
20.根据权利要求19所述的药品或药物组合物,其特征在于,其包含也起抗感染作用的至少一种另外的活性成分。
21.根据权利要求19或20所述的药品或药物组合物,其特征在于,其包含对除细菌感染以外的其他状况有效的至少一种另外的活性成分。
说明书
本发明涉及一种制备聚胍的新方法、根据该方法制备的缩聚产物及其作为抗微生物剂或抗感染剂的用途。
现有技术
长久以来就已公知由下述通式表示的聚胍及其不同的衍生物。
专利文献早在1943年就在美国专利2 325 586中描述了通过i)胍或胍盐、ii)卤化氰、iii)二氰胺、或iv)异腈二卤化物与二胺的缩聚,或v)两种双氰胺彼此的缩聚(产生氰基取代的聚胍)得到多种聚胍的多种制备方法,以及将生成的聚胍作为染色助剂的用途:
早在那时,亚烃基二胺和亚苯基二胺以及稍后也公知为 的氧化烯二胺或聚醚二胺就已公开为反应i)至iv)中使用的二胺。
数十年后,此类聚胍也被证实是优良的杀生物剂。因此,以Oskar Schmidt为中心的团队在WO 99/54291 A1中公开了杀微生物的聚六亚甲基胍的制备,在WO 01/85676 A1中公开了通过缩合胍和聚氧化烯来制备杀生物的聚胍,并且还在WO 2006/047800 A1中公开了充当杀生物剂,特别是充当杀真菌剂的聚胍衍生物,它们是通过将胍与亚烃基二胺和氧化烯二胺的混合物缩聚来形成且具有的毒性低于那些仅含有两种类型的二价残基(Rest)R1中的一种的聚合物。
在WO 02/30877 A1中描述了作为消毒剂的类似聚胍,其在链中包含另外的亚苯基部分。一个俄罗斯研究小组(Tets,Tets和Krasnov)在从其中派生出US 2011/0269936 A1和EP 2.520.605 A1的WO 2011/043690 A1中公开了如下化学式的杀生物剂聚胍,该聚胍是在存在水合肼的情况下通过缩聚胍和己二胺制备而成:
因此,肼在缩聚过程中-至少在形式上-取代仅一个或两个胍基中的氨基,由此得到嵌段共聚物(Block-Copolymere),其中聚(六亚甲基胍)嵌段与聚(六亚甲基氨基胍)嵌段交替,并且这两种类型的嵌段分别通过胍二聚体相互连接,如下所示:
这种聚合物和它们的酸式盐也可用作对抗细菌、病毒和真菌的杀生物剂。然而在制备了7种不同的聚合物的这些申请的实施例中,除了指明实施例1中的聚合物为“固体的,几乎无色的,透明的物质”外,对所得产物没有发现任何物理数据。
关于胍与二胺缩聚过程中能够产生的可能结构,有几篇文章出自格拉茨理工大学(Graz University of Technology)的研究团队,例如Albert等人,Biomacromolecules 4(6),1811-1817(2003),和Feiertag等人,Macromol.Rap.Comm.24(9),567-570(2003)。除了用初始单体(Ausgangsmonomere)中的一种终止线性聚合物链的若干的可能性外,通常在部分地依赖于二胺的链长的不可忽略的部分中也会形成以下通式的环状分子:
实际上所有上述聚胍衍生物的主要缺点首先在于产物不可忽视的毒性,和–若使用高反应活性成分-制备方法相对昂贵,以及使用从毒理学方面众所周知的有问题的成分,例如肼(Hydrazin),因此本发明人已经开始研究问题的解决方案。
在其研究过程中发明人发现,氨基胍和二氨基胍与胺的缩聚产物比上述引用文献WO 2011/043690 A1,US 2011/0269936 A1和EP 2.520.605 A1中含有胍的结构相近的缩聚物具有令人吃惊的明显更低的毒性,但也是有效的抗微生物物质。
未决专利申请AT A 53/2013和PCT/AT2014/050026中公开了这些结果,在这些申请中要求保护如下化学式的聚胍衍生物及其盐:
其中
X选自-NH2,氨基胍基和1,3-二氨基胍基;
Y选自-H和-R1-NH2;
或X与Y一起代表化学键,以便得到环状结构;
R1选自包含2至20个碳原子的二价有机残基,其中一个或更多个碳原子任选地被O或N替换;
a和b各自是0或1,其中当不含有1,3-二氨基胍单元时,a+b≠2;
R2选自-H和-NH2,
其中当a+b=0时,R2是-NH2,
当a+b=1时,R2是-H或-NH2,并且
当a+b=2时,R2是-H,且
n≥2。
类似于以往已知的现有技术,将相应的二胺与氨基胍和/或二氨基胍通过加热进行缩聚来作为制备这种新的聚(二)氨基胍的方法。
发明人不希望被理论所束缚,假定氨基胍部分和二氨基胍部分(只要上下文中没有其他的内容,则以下共同地称为“氨基胍”)比胍基,特别是比含有上述肼撑桥连的胍二聚体的那些聚合物更被人类的真核细胞耐受。
然而这些新的氨基胍化合物中的某些在其抗微生物有效性或毒性方面不能证实是完全令人满意的,并且由于某些二胺的使用需要非常高的温度以用于熔融聚合并且有时仍会带来有问题的剩余单体含量,所以制备方法也是需要改进的。
因此,本发明的目的是提供具有更好性质的另外的聚胍衍生物,以及所述聚胍衍生物的有利的制备方法。
发明内容
本发明的这一目标一方面通过提供一种由胍、氨基胍或二氨基胍G与一种或更多种苯甲基衍生物或烯丙基衍生物BA根据如下反应方案获得缩聚产物的制备方法来实现:
其中
X各自独立地代表离去基团;
R1各自独立地代表具有至少一个芳香环的芳香族环体系,所述芳香环任选地包含一个或更多个选自O、N和S的杂原子且任选地被一个或两个乙烯基团取代,-CH2-X基团被结合至所述乙烯基,或R1各自独立地代表乙烯基;
Gua代表胍二基(guanidindiyl)、氨基胍二基(aminoguanidindiyl)或二氨基胍二基(diaminoguanidindiyl);
Y代表H-Gua,并且
Z代表H;或
Y和Z一起代表化学键,以便得到环状结构;
其中使至少一种苯甲基衍生物或烯丙基衍生物BA与过量的胍、氨基胍或二氨基胍G在消除HX的情况下发生缩聚反应,以便得到对应于如下化学式(I)、(II)或(III):
或具有通过在消除相应的胍后的环化得到的环状结构的聚胍,或所述聚胍的盐。
不同于现有技术,在此制备方法中,缩聚并非在裂解氨的情况下,而是在裂解离去基团X的情况下实现,裂解离去基团X优选地以卤化氢例如如HCl或HBr,或磺酸例如CH3SO2OH(MsOH)的形式,其与存在于分子中的氨基或亚氨基形成酸加成盐,这进而使得不必使用除酸剂。
此外,这还会导致缩聚不一定在熔融条件下进行,尽管由于方法经济性的原因,根据本发明熔融聚合是优选的反应路线。因此,在优选的实施方案中,通过将反应物加热到其熔融温度以上的温度来使至少一种苯甲基衍生物或烯丙基衍生物BA与胍、氨基胍或二氨基胍G反应,其中聚合反应优选地被进行持续至少2个小时,更优选地至少3个小时。特别地,与发明人之前的方法类似,反应分两步在不同的温度下进行,即第一步在较低温度下,第二步在较高温度下进行,以便保证尽可能完全转化并由此保证在减少残余单体含量的同时链长更长。
然而,令人吃惊的是,本发明人发现,使用苯甲基或烯丙基结构产生具有不同于从现有技术中已知的那些的结构的缩聚产物的混合物。然而,主要产物与发明人之前的申请中公知的各自具有仅单取代的氮的结构不相对应,而且早已单取代的氮明显优选地进行第二次反应,以便得到化学式(I)至(III)的上述结构。
不希望被理论所束缚,发明人假定在苯甲基或烯丙基的亚甲基上发生亲核取代时归因于过渡态的内消旋稳定化(Mesomeriestabilisierung)的反应物的反应性与最初所形成的单取代氮加合物增加的反应性一起导致了所得的氮双取代,从而进一步假定,确定至少大多数的已知的苯甲基或烯丙基结构也有类似的结果,也就是说,在带有一个连接在芳香环或双键上的亚甲基的结构,或其组合(即在肉桂基结构的情况下)中有类似的结果,其中-涉及到苯甲基残基时-显然已知的插烯原理起作用(参见下文的实施例8)。后者当然还可用在脂肪族残基中的共轭双键上,例如在丁二烯代替乙烯这种情况中。基于这一原因,目前不应该特别地限制芳香环和双键上可能的其他取代基,只要由此并不消除各环的芳香性,或芳香环中或双键上的电子密度不发生巨大改变,特别是在互变异构效应的情况下,例如酮/烯醇,亚胺/烯胺等。
在化学式(1)至(3)给出的结构中,胍基单元或氨基胍单元通过两次连接在链中的氮原子被定位在链的外部,其中化学式(I),(II)或(III)的结构类型与大多数形成的低聚物种类的光谱学证据相一致。
借助HMBC-NMR来确定所得到的新聚胍的连接类型:因此例如在实施例1中的聚氨基胍中,通过氮连接的苯甲基CH2-质子(3.8和4.2ppm时的AB体系)的相应远程耦合可通过分别连接在低聚物链中的N原子以及两个苯甲基碳原子(64ppm时)得以证实。此外,还找到了与另一个超出亚氨基官能性的胍氮的苄基化相关的信号作为高度支化副组分(依据1H-NMR,~15%)的指征(4.3和4.5ppm时的AB体系,160ppm时胍碳区中的HMBC远程信号)。苯甲基亚氨基官能性的另一组NMR信号(以8ppm的1H-位移,以150ppm的13C-位移)与索美勒氧化类型的低聚物悬垂物相关联,这与质谱数据(对于所有低聚物的m/z[M-2]类的双峰)相一致。
发明人甚至还期待这种新的结构类型具有比其早期的聚氨基胍更好的抗微生物活性,这也能够被证实,如下文中的本发明的实施例示出的那样:杀生物活性明显提高,同时毒性还有所降低。
发明人不希望受到理论的限制而假定,后者可能应归因于相对于以前的聚氨基胍有更长的平均链长,并且剩余单体含量更少。
为了在反应时间、链长和剩余单体含量之间寻找到尽可能好的折中方案,以此来优化反应条件,发明人开展了具有不同的苯甲基或烯丙基衍生物BA与胍G的比例、不同温度以及不同反应时间的一系列实验,并发现当BA/G比例刚好低于2时所得产物具有最好的生物结果,其中优选首先应该将反应混合物在150-170℃左右的温度下加热持续2至3小时,并且接着在180-190℃的温度下加热持续1至2小时。
本发明的第二方面在于,通过本发明提供相应于下列化学式(I)至(III)的新聚胍,即对应于下述化学式(I)的聚胍:
或具有通过在消除胍后的环化得到的环状结构的聚胍;
对应于下述化学式(II)的聚胍:
或具有通过在消除氨基胍后的环化得到的环状结构的聚胍;以及
对应于下述化学式(III)的聚胍:
或具有通过在消除二氨基胍后的环化得到的环状结构的聚胍;
其中,R1代表具有至少一个芳香环的芳香族环体系,所述芳香环任选地包含一个或更多个选自O、N和S的杂原子且任选地被一个或两个乙烯基取代,-CH2-X基团被结合至所述乙烯基,或R1代表乙烯基;并且在优选的实施方案中R1选自任选地被取代的苯、二乙烯基苯、呋喃、吡咯、噻吩、吡啶、联苯、芴和乙烯的二价残基,更优选地选自已提供了很好的结果的苯、二乙烯基苯、吡啶、联苯和乙烯的二价残基。
由于新结构的高抗微生物效果,本发明在第三方面提供了一种上文定义的新聚胍用作抗生素和抗感染药,优选用于对抗人类或动物患者中的细菌、病毒和真菌感染。聚胍在此能够用于局部施用或全身施用,优选用于以药品或药物组合物的形式施用。
但作为另一种选择,新聚胍还能够用作体外抗微生物剂,优选用作抗微生物涂料、涂层、箔或膜或其他类似物的活性组分。
本发明在第四方面提供了一种用于对抗人类或动物患者中的细菌、病毒和真菌感染的药品或药物组合物,其包含至少一种新聚胍作为抗感染药,此外还优选包含至少一种药学上可接受的载体或赋形剂,并且任选地具有一种或更多种佐剂和/或一种或更多种另外的活性剂。
优选地,为了增强效果并利用可能的协同作用,药品或药物组合物包含也具有抗菌作用的至少一种另外的活性成分。所述至少一种另外的活性成分在此有时也对除细菌感染以外的其他状况有效。止泻剂和所谓的胃保护剂仅提及作为实例。
实施例
实施例1
制备聚氨基胍(1)
将α,α’-二氯-对二甲苯(880mg,5.03mmol)和1.95份相对应的化学计量的氨基胍盐酸盐(1083mg,9.80mmol)在敞开的反应器中首先搅拌加热至160℃持续3小时,然后搅拌加热至180℃持续2小时。当反应混合物冷却至低于80℃之后,向反应产物中加入十倍量的水,并且借助搅拌或超声波处理完全混合后获得带有固体成分痕迹的清澈淡黄色溶液。溶液经0.2μm的PFTE膜过滤,然后再蒸发,以便得到黄色的无定形固体聚胍(1)。
为了进行分析,将样品溶解在十倍量的D2O中。在记录1H-和13C-NMR谱时,加入DSS(4,4-二甲基-4-硅戊烷-1-磺酸)作为内标以供参考:
1H NMR(D2O),δ(ppm):3.72-3.91(ad,CH2A-N(Gua)-CH2A,JA,B=12.4Hz,CH2A链),3.93-4.05(as,CH2-NH-Gua,CH2端),4.10-4.23(ad,CH2B-N(Gua)-CH2B,JA,B=12.4Hz,CH2B链),4.29-4.39(m,CH2Aα-Gua),4.45-4.52(m,CH2Bα-Gua),7.30-7.83(m,=CH Ar),8.08(as,N=CH)。
13C NMR(D2O),δ(ppm):46.25,46.56,46.94(CH2α-Gua),56.90,56.97,57.03(CH2端),63.87,64.02(CH2-N(Gua)-CH2链),128.93,129.04,129.57,129.63,129.78,129.84,130.20,130.32,130.49,130.66,132.10,132.17,132.30,132.40,132.62,132.67,132,75,132.83,132.92,133.20(CH Ar),135.02,135.19,137.54,137.92,138.13,138.50,139.07,139.23,141.31,142.53(CqAr),150.21,151.05,151.12(N=CH),157.60,159.67,159.73,160.85(CqGua)。
在3.72-3.91ppm和4.10-4.23ppm(1H-轴)范围中以及在64.02ppm(13C-轴)时的NMR信号证实了氨基胍的两次取代的氮原子的存在。
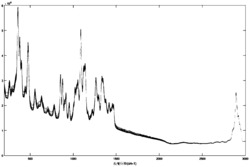
MALDI-MS–MALDI-TOF(在正离子模式中(基质抑制关闭));扫描20-3000m/z(偏转关闭);基质:ACH(α-氰基-4-羟基肉桂酸);(m/z):247.3,249.3,251.4,425.3,427.3,601.4,603.4,777.5,779.5,953.7,955.7,1129.8,1131.9,1306.0,1308.0,1482.1,1484.1,1658.0,1660.0,1834.1,1836.1,2010.2,2012.2,2186.3,2188.3,2362.4。
实施例2
制备聚氨基胍(2)
用与实施例1相似的方式,由α,α’-二氯-间二甲苯和氨基胍盐酸盐得到黄色的无定形的完全溶解于水的固体聚胍(2)。
1H NMR(D2O),δ(ppm):3.73-3.92(ad,CH2A-N(Gua)-CH2A,JA,B=12.7Hz,CH2A链),3.94-4.05(as,CH2-NH-Gua,CH2端),4.10-4.23(ad,CH2B-N(Gua)-CH2B,JA,B=12.7Hz,CH2B链),4.29-4.38(m,CH2Aα-Gua),4.45-4.53(m,CH2Bα-Gua),7.23-7.85(m,=CH Ar),8.10(as,N=CH)。
13C NMR(D2O),δ(ppm):46.36,46.66,47.01(CH2α-Gua),57.01,57.04,57.12,57.14(CH2端),63.94(CH2-N(Gua)-CH2链),129.63,129.75,130.09,130.20,130.83,131.38,131.44,131.53,131.57,131.67,131.82,131.89,132.18,132.34,132.73,133.52,134.23,134.52,135.29(CH Ar),135.72,135.81,136.12,138.59,138.69,138.73,139.13,139.77,139.90,140.30(CqAr),151.24(N=CH),157.67,159.78,159.81,160.86(CqGua)。
在3.73-3.92ppm和4.10-4.23ppm(1H-轴)范围中以及在63.94ppm(13C-轴)时的NMR信号再次证实了氨基胍的两次取代的氮原子的存在。
MALDI-MS–MALDI-TOF(m/z):247.3,249.3,251.4,425.3,427.3,601.4,603.4,777.5,779.5,953.7,955.7,1129.8,1131.9,1306.0,1308.0,1482.1,1484.1,1658.0,1660.0,1834.1,1836.1,2010.2,2012.2,2186.3,2188.3。
实施例3
制备聚氨基胍(3)
用与实施例2类似的方式,由132mg(0.5mmol)α,α’-二溴-间二甲苯(代替二氯衍生物),以及1.75份化学计量的氨基胍盐酸盐(97mg,0.88mmol)得到褐色的无定形的水溶性固体聚胍(3)。
1H NMR(D2O),δ(ppm):3.63-3.95(m,CH2A-N(Gua)-CH2A,CH2A链),3.95-4.08(as,CH2-NH-Gua,CH2端),4.13-4.24(ad,CH2B-N(Gua)-CH2B,JA,B=12.5Hz,CH2B链),4.31-4.40(m,CH2Aα-Gua),4.47-4.55(m,CH2Bα-Gua),7.17-7.86(m,=CH Ar),8.12(as,N=CH)。
13C NMR(D2O),δ(ppm):46.38,46.64,46.99(CH2α-Gua),56.98,57.11,57.48(CH2端),63.90(CH2-N(Gua)-CH2链),128.58,129.08,129.64,129.76,130.05,130.20,130.81,130.98,131.35,131.41,131.51,131.71,131,80,131.87,132.16,132.33,132.69,133.49,134.21,134.51,135.29(CH Ar),135.66,135.76,136.06,138.68,138.98,139.07,139.25,139.72,139.85,140.25(CqAr),150.46,151.29(N=CH),159.73,160.84(CqGua)。
在3.63-3.95ppm和4.13-4.24ppm(1H-轴)的范围中以及在63.90ppm(13C-轴)时的NMR信号再次证实了氨基胍的两次取代的氮原子的存在。
MALDI-MS–MALDI-TOF(m/z):247.3,249.3,251.4,425.3,427.3,601.4,603.4,777.5,779.5,953.7,955.7,1129.8,1131.9,1306.0,1308.0,1482.1,1484.1,1658.0,1660.0,1834.1,1836.1,2010.2,2012.2,2186.3,2188.3。
实施例4
制备聚二氨基胍(4)
用与实施例2类似的方式,由88mg(0.5mmol)α,α’-二氯-间二甲苯以及1份化学计量的二氨基胍盐酸盐(68mg,0.5mmol)得到黄色的无定形的水溶性固体聚胍(4)。
借助1H-和13C-NMR进行的结构分析表明,除了存在实施例1至3中发现的产物种类外,还存在较大份额的高支化度组分,其中超出亚氨基官能性的另一胍氮被苄基化。
实施例5
制备聚胍(5)
用与实施例3类似的方式,由132mg(0.5mmol)α,α’-二溴-对二甲苯以及1.75份化学计量的胍盐酸盐(83mg,0.88mmol)得到水溶性的红色无定形固体聚胍(5)。
借助1H-和13C-NMR进行的结构分析表明,除了存在实施例1至3中发现的产物种类外,还存在较大份额的高支化度组分,其中超出亚氨基官能性的另一胍氮被苄基化。
MALDI-MS–MALDI-TOF(m/z):355.3,382.3,516.4,543.4,677.3,704.5,838.5,865.5,999.6,1026.6,1160.7,1187.7,1321.8,1348.8,1483.9,1510.9,1672.0,1833.1,1995.2。
实施例6
制备聚氨基胍(6)
将2,6-双(溴甲基)吡啶(265mg,1mmol)和1.95份化学计量的氨基胍盐酸盐(216mg,1.95mmol)在敞开的反应器中首先搅拌加热至160℃持续1.5小时,然后搅拌加热至180℃持续1.5小时。当反应混合物冷却至低于80℃之后,向反应产物中加入水(4.81ml),并且在借助搅拌或超声波处理完全混合以及通过0.2μm的PFTE膜过滤后,得到清澈的深褐色溶液。
MALDI-MS–MALDI-TOF(m/z):248.4,250.4,252.4,421.4,423.4,425.4,427.4,429.4,598.4,600.4,602.4,604.4。
实施例7
制备聚氨基胍(7)
用与实施例2类似的方式,由4,4’-双(氯甲基)联苯(251mg,1mmol)以及1.95份化学计量的氨基胍盐酸盐(216mg,1.95mmol)得到黄色的无定形的固体聚胍(7),其除了少量固体残留物之外易溶于水。
MALDI-MS–MALDI-TOF(m/z):323.4,325.4,327.4,575.4,577.4,579.4,827.6,829.6,831.6。
实施例8
制备聚氨基胍(8)
8.1制备3,3'-(1,3-亚苯基)-(2E,2'E)-二丙烯酸二甲酯
在排除空气的条件下,向将0.75mmol间苯二醛放入10ml THF中构成的溶液中加入将2.05份化学计量的(甲氧基羰基亚甲基)三苯基膦放入15ml THF中构成的溶液。将反应混合物在50℃下搅拌持续12小时并浓缩。色谱提纯(二氧化硅、二氯甲烷)得到0.62mmol白色固体(83%理论(d.Th.))。
1H NMR(400MHz,CDCl3):3.82(s,6H),6.47(d,J=16Hz,2H),7.42(dd,J=7.7+7.7Hz,1H),7.54(dd,J=7.7+1.7Hz,2H),7.64(t,J=1.7Hz,1H),7.69(d,J=16Hz,2H)。
8.2制备(2E,2'E)-3,3'-(1,3-亚苯基)-双(丙-2-烯-1-醇)
在施伦克反应器中,将1.50mmol 3,3’-(1,3-亚苯基)-(2E,2’E)-二丙烯酸二甲酯溶解在30ml无水二氯甲烷中。在-78℃下缓慢滴加作为在甲苯(6.75ml)中的1M溶液的4.5份化学计量的二异丁基氢化铝。将反应混合物在-78℃下搅拌2小时,并且接着在0℃下用甲醇水解。过滤产生的白色沉淀,将滤液浓缩并色谱提纯(二氧化硅,DCM:EE 1:1),其中分离出1.05mmol(70%理论)的白色固体。
1H NMR(400MHz,CDCl3)δ(ppm):4.26(m,4H),6.33(dm,J=16Hz,2H),6.56(br,d,J=16Hz,2H),7.22(m,3H),7.34(br s,1H)。
8.3制备1,3-双((E)-3-氯丙-1-烯-1-基)苯
在施伦克反应器中,将0.95mmol 3,3'-(1,3-亚苯基)-(2E,2'E)-二丙烯酸二甲酯和3份化学计量的二异丙基乙胺(DIPEA,2.85mmol)加入到20ml二氯甲烷中并冷却至-40℃,然后加入2.38mmol甲磺酰氯,将反应混合物在室温下搅拌12小时。在除去溶剂后,色谱提纯(二氧化硅,DCM)粗产物,其中分离出0.57mmol(60%理论)的白色结晶固体。
1H NMR(400MHz,CDCl3)δ(ppm):4.25(dd,J=7.1+1.2Hz,4H),6.34(dt,J=15.7+7.1Hz,2H),6.65(dt,J=15.7+1.2Hz,2H),7.30(m,3H),7.40(m,1H)。
8.4制备聚氨基胍(8)
用与实施例2类似的方式,由1,3-双((E)-2-氯乙烯基)苯(200mg,1mmol)以及1.95份化学计量的氨基胍盐酸盐(216mg,1.95mmol)得到黄色的半透明的水溶性凝胶聚胍(8)。
MALDI-MS–MALDI-TOF(m/z):303.3,531.4,759.6,833.7,987.8,1061.9,1216.0.
实施例9
制备聚氨基胍(9)
9.1制备顺式-1,4-双(甲磺酰氧基)-2-丁烯
将10g顺式-2-丁烯-1,4二醇(113mmol)与3.0份化学计量的二异丙基乙胺(44g,340mmol,60ml)溶解在250ml二氯甲烷中,并在氩保护气氛中冷却至-40℃,然后逐份地加入2.4份化学计量的甲磺酰氯(30.9g,270mmol,20.9ml),并且将反应混合物在1小时内加温至+10℃。将澄清的黄色溶液倾倒至500ml冰水中,再用500ml冰水、接着用200ml 2N HCl、然后两次分别用200ml饱和NaHCO3溶液、最后再两次分别用200ml水来清洗有机相。通过Na2SO4来干燥产物的二氯甲烷溶液,将溶剂在真空中除去直至出现白色沉淀,之后加入最少量的二氯甲烷以便重新得到清澈的溶液。加入25ml乙醚后,在-20℃下使产物从溶液中结晶,之后分离出10g结晶的白色固体顺式-1,4-双(甲磺酰氧基)-2-丁烯。
1H NMR(400MHz,CDCl3)δ(ppm):3.04(s,3H),4.84(m,2H),5.95(m,1H)。
9.2制备聚氨基胍(9)
在氩气保护气氛下,将顺式-1,4-双(甲磺酰氧基)-2-丁烯(246mg,1mmol)与1.95份化学计量的氨基胍盐酸盐(216mg,1.95mmol)在封闭的反应器中边搅拌边加热,首先加热至160℃持续3小时,之后加热至180℃持续2小时。在反应混合物冷却至80℃以下后,向反应产物中加入水(4.67ml),并得到清澈的黄红色溶液。
MALDI-MS–MALDI-TOF(m/z):201.3,251.3,253.3,297.2,325.3,327.3,349.2,377.3,423.3,451.3,453.3,519.3。
实施例10
制备聚氨基胍(10)
在氩气保护气氛下,将1,4-二氯-2-丁烯(262mg,1.3mmol)与1.95份化学计量的氨基胍盐酸盐(216mg,1.95mmol)在封闭的反应器中边搅拌边加热,并且重复(每小时三次)更换新鲜氩气,首先加热至150℃持续2小时,之后加热至170℃持续1小时。在反应混合物冷却至80℃后向反应产物中加入水(4.67ml),并得到清澈的黄红色溶液。
MALDI-MS–MALDI-TOF(m/z):201.3,251.3,253.3,297.2,325.3,327.3,377.3,423.3,451.3,453.3。
比较例1
由二胺和氨基胍制备聚氨基胍
将23mmol的1,3-二氨基胍盐酸盐和24mmol的4,9-二氧杂十二烷-1,12-二胺放在用干燥管闭合的反应器中加热至120℃持续90分钟,同时搅拌,然后将温度升高至180℃再加热100分钟,并且在反应时间还剩下45分钟时减压(50mbar)。将反应混合物冷却到低于80℃之后,向凝胶状反应产物中加入25ml水。数小时后得到澄清的溶液。
将所得水溶液样品中的水蒸发掉,并在真空中将所得剩余物干燥,这时得到红色的粘稠液体。将该液体溶解在2ml D2O(氘化程度>99.5%)中,并且记录下1H-核磁共振(1H-NMR-)谱。可用这种方式区分的、产物中剩余物R1的亚甲基质子组的位置如下:
1H NMR(D2O),δ(ppm):1.54-1.67(m,OCH2CH2CH2CH2O),1.80-1.95(m,NCH2CH2),3.23-3.38ppm(m,NCH2),3.42-3.65ppm(m,CH2CH2OCH2CH2)。
这证实了所用二胺组分4,9-二氧杂十二烷-1,12-二胺的结构。
实施例11
活性测定:抗微生物/抗真菌/抗病毒作用
在多次实施的筛选系统中测试新化合物的活性。借助MHK测试来研究抗菌和抗真菌活性。MHK是指“最小抑菌浓度”(英文:MIC“minimal inhibitory concentration”)并且表明了物质的最低的浓度,在此浓度下仅凭裸眼无法辨别微生物的繁殖。通过所谓的滴定法来确定MHK,其中先将物质稀释后再添加病原体。
通常以如下方式确定抗生素的浓度,即该浓度正好抑制菌株的生长。MHK用微克每毫升(μg/ml)或用体积百分比(Vol.-%)表示,通常以log2梯度(log2-Schritten)实现稀释。在这里将1%的初始浓度每次稀释两倍,从而得到0.5%、0.25%、0.125%等的测试浓度。为此,该值越低反映了抗感染药的活性更佳。
按照EUCAST(European Committee for Antimicrobial Susceptibility Testing抗微生物剂敏感性测试欧洲委员会)所要求的标准,并根据欧洲临床微生物学与感染性疾病学会(ESCMID)的AFST-(“Antifungal Susceptibility Testing”抗真菌药敏测试)规定来进行上述测试。
病毒的筛选系统是感染系统,其中在体外感染宿主细胞,并在未感染前或感染后加入测试物质并确定其活性。所有上述测试均依照海洋生物制药企业(SeaLife Pharma)内部关于药品筛选的标准规定来实施,其中采用类似于抗菌/抗真菌测试中相似的稀释系列。
在下页表1至3中给出了实施例1、3、4和5以及比较例1中依据本发明的新化合物对若干多重耐药的细菌和真菌以及病毒的抗感染效果的测试结果。这些数据分别为多次测试的平均值。
很显然,依据本发明的新化合物既对革兰氏阳性病原体也对革兰氏阴性病原体表现出优异的活性。
实施例12
毒性测试
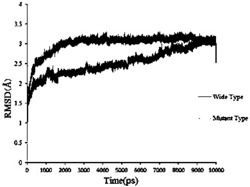
此外,从所附图1还可知,依据本发明的新聚胍在具有极佳的抗微生物活性的各个浓度上同时表现出极低的毒性,正如作为Y轴上细胞模型的暴露的HaCaT-细胞系的存活细胞的比例所清晰显示出来的那样。
制备聚胍的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。















动态评分
0.0