

IPC分类号 : B01J27/06,C01B32/194,C01G33/00,B82Y30/00,B82Y40/00









专利摘要
本发明公开了一种Nb3O7F纳米阵列/石墨烯异质结复合材料的制备方法,包括如下步骤:配置石墨烯水溶液,搅拌后进行超声剥离,使石墨烯形成均匀分散液;向上述分散液中加入氢氟酸,搅拌并辅以超声,使得剥离的石墨烯表面被充分刻蚀,形成碳氟键;称取NbCl5粉末,加入到上述溶液中,搅拌充分后再添加氢氟酸,继续搅拌充分;将上述溶液转移至特氟龙内衬的反应釜中进行水热反应;反应结束后,将产物离心分离,并用去离子水和无水乙醇清洗,在烘箱中干燥;将干燥的产物热处理,以除去有机物,最终得到Nb3O7F纳米阵列/石墨烯异质结复合材料。本发明过程简单,操作条件易于控制,实现了低温下制备石墨烯基异质结材料。
权利要求
1.一种Nb3O7F纳米阵列/石墨烯异质结复合材料的制备方法,其特征是:包括如下步骤:
(1)配置石墨烯水溶液,搅拌后进行超声剥离,使石墨烯形成均匀分散液;
(2)向上述分散液中加入氢氟酸,搅拌并辅以超声,使得剥离的石墨烯表面被充分刻蚀,形成碳氟键;
(3)称取NbCl5粉末,加入到上述溶液中,搅拌充分后再添加氢氟酸,继续搅拌充分;
(4)将上述溶液转移至特氟龙内衬的反应釜中进行水热反应;
(5)反应结束后,将产物离心分离,并用去离子水和无水乙醇清洗,在烘箱中干燥;
(6)将干燥的产物热处理,以除去有机物,最终得到Nb3O7F纳米阵列/石墨烯异质结复合材料。
2.根据权利要求1所述的Nb3O7F纳米阵列/石墨烯异质结复合材料的制备方法,其特征是:所述步骤(1)中,配置石墨烯水溶液所采用的石墨烯为纳米片结构,石墨烯的添加量,是按质量百分比,以Nb3O7F为基准,添加量为0.5~3.0wt.%。
3.根据权利要求1所述的Nb3O7F纳米阵列/石墨烯异质结复合材料的制备方法,其特征是:所述步骤(1)中,搅拌时间为1~3h,超声剥离时间为5~10h。
4.根据权利要求1所述的Nb3O7F纳米阵列/石墨烯异质结复合材料的制备方法,其特征是:所述步骤(1)中,石墨烯水溶液中添加有表面活性剂,所述表面活性剂为三嵌段表面活性剂,表面活性剂在步骤(4)的反应溶液中的浓度控制在0~40g/L。
5.根据权利要求1或4所述的Nb3O7F纳米阵列/石墨烯异质结复合材料的制备方法,其特征是:所述步骤(1)中,石墨烯水溶液中添加有络合剂,所述络合剂为一水合柠檬酸,络合剂在步骤(4)的反应溶液中的浓度控制在0~40g/L。
6.根据权利要求1所述的Nb3O7F纳米阵列/石墨烯异质结复合材料的制备方法,其特征是:所述步骤(2)中,加入氢氟酸后的溶液中,氢氟酸的浓度控制在0.1~0.15mol/L;所述步骤(3)中,再次加入氢氟酸到溶液中,最终氢氟酸浓度控制在0.3~0.4mol/L。
7.根据权利要求1所述的Nb3O7F纳米阵列/石墨烯异质结复合材料的制备方法,其特征是:所述步骤(2)中,搅拌时间为5~15min,超声剥离时间为10~30min;所述步骤(3)中,搅拌时间为2~6min。
8.根据权利要求1所述的Nb3O7F纳米阵列/石墨烯异质结复合材料的制备方法,其特征是:所述步骤(4)中,水热温度为120~200℃,水热时间为12~48h。
9.根据权利要求1所述的Nb3O7F纳米阵列/石墨烯异质结复合材料的制备方法,其特征是:所述步骤(5)中,去离子水清洗为常温清洗,洗涤次数为3~5次;乙醇清洗为常温清洗,洗涤次数为1~3次;干燥温度为60~80℃,干燥时间大于5h。
10.根据权利要求1所述的Nb3O7F纳米阵列/石墨烯异质结复合材料的制备方法,其特征是:所述步骤(6)中,热处理温度为400℃,热处理时间为2~4h。
说明书
技术领域
本发明涉及一种石墨烯基异质结材料的制备方法,具体是一种Nb3O7F纳米阵列/石墨烯异质结材料的制备方法。
背景技术
近年来,新型半导体光催化材料的快速发展,为降解有机污染物提供了新的选择方式;同时,一些新型半导体光催化材料能突破传统TiO2光催化材料存在的限制,尤其是载流子复合速率高、光谱响应低等问题,从而引起了学者的广泛关注。最近,Nb3O7F 因与TiO2的电子结构和能带结构相似,结晶性好、物相单一、化学惰性优异、抗氧化能力强、光催化活性高、载流子复合速率低等优点,被认为是一种很有前途的光催化材料。然而,Nb3O7F在光催化应用中存在电荷传输速率慢、量子效率低以及吸附性能差等缺点,致使其光催化效率得不到进一步提升。因此,寻找一种既能优化Nb3O7F的量子效率,又能增加载流子传输速率和吸附性能的途径,是实现其光催化应用的关键。
石墨烯是由单层碳原子组成的二维层状材料,具有优异的导电、导热性能,其较大的比表面积使其具有较强的吸附特性,是一种很好的电子传输材料。石墨烯与半导体材料形成异质结,可优化界面结构,加快半导体的载流子传输速率,减少载流子复合几率,从而提高量子效率,同时还可利用其大的比表面积优势显著增强吸附性能。然而,石墨烯纳米片易于再聚集,不易分散,从而失去石墨烯的特有功能;而且,石墨烯表面惰性强,很难制备理想的石墨烯基异质结材料。尽管人们开始尝试采用不同方法来制备石墨烯基异质结材料,但Nb3O7F纳米阵列/石墨烯异质结的文献或专利还未曾有报道。
文献1(Peifang Wang,Materials Letters,2013,101,41-43)采用溶剂热法,直接将还原氧化石墨烯沉积在TiO2阵列上,并研究了其光催化性能。该方法只是将石墨烯简单地沉积在TiO2阵列上,未形成良好的异质结构,结果光催化性能的提升幅度有限。
文献2(Yu Zhao,Scientific Reports,2016,6,32327)采用水热法直接在还原氧化石墨烯薄膜上生长ZnO纳米棒,从而形成ZnO/石墨烯异质结。该方法操作过程繁琐,成本较高,氧化石墨烯的还原程度较难控制,很难完全还原,极大地影响了产物的性能。
文献3(Qisheng Wang,Advanced Materials,2016,28,6497-6503)采用CVD在高温下将PdS沉积在石墨烯上,制成Graphene/PdS异质结,并研究了其伏安特性。该方法采用物理法,设备昂贵,成本较高,且工艺复杂,制备的异质结均匀性不易控制。
文献4(Chemistry-An Asian Journal,2016,11,584-595)采用光致还原法,在还原氧化石墨烯的同时,原位形成Ag3PO4/rGO异质结材料,并研究了其光催化性能。该方法中使用的氧化石墨烯还原程度低,缺陷较多,光催化性能得不到有效提升。
发明内容
本发明的目的是提供一种Nb3O7F纳米阵列/石墨烯异质结的制备方法,以解决现有技术中存在的问题,如直接使用石墨烯为基质时其在水溶液中难以稳定分散,表面不易被共价,异质结生长困难的难点;而直接使用氧化石墨烯为基质时其还原程度低,性能无法提升,以及异质结均匀性不好等问题。
技术方案:为实现上述目的,本发明采用的技术方案为:
一种Nb3O7F纳米阵列/石墨烯异质结复合材料的制备方法,包括如下步骤:
(1)配置石墨烯水溶液,搅拌后进行超声剥离,使石墨烯形成均匀分散液;
(2)向上述分散液中加入氢氟酸,搅拌并辅以超声,使得剥离的石墨烯表面被充分刻蚀,形成碳氟键;
(3)称取NbCl5粉末,加入到上述溶液中,搅拌充分后再添加氢氟酸,继续搅拌充分;
(4)将上述溶液转移至特氟龙内衬的反应釜中进行水热反应;
(5)反应结束后,将产物离心分离,并用去离子水和无水乙醇清洗,在烘箱中干燥;
(6)将干燥的产物热处理,以除去有机物,最终得到Nb3O7F纳米阵列/石墨烯异质结复合材料。
所述步骤(1)中,配置石墨烯水溶液所采用的石墨烯为纳米片结构,石墨烯的添加量,是按质量百分比,以Nb3O7F为基准,添加量为0.5~3.0wt.%。
所述步骤(1)中,搅拌时间为1~3h,超声剥离时间为5~10h。
所述步骤(1)中,石墨烯水溶液中添加有表面活性剂,所述表面活性剂为三嵌段表面活性剂,表面活性剂在步骤(4)的反应溶液中的浓度控制在0~40g/L。
所述步骤(1)中,石墨烯水溶液中添加有络合剂,所述络合剂为一水合柠檬酸,络合剂在步骤(4)的反应溶液中的浓度控制在0~40g/L。
本发明中,氢氟酸采用两次添加,第一次添加在步骤(2)中,目的是为了石墨烯的表面刻蚀,从而形成碳氟键,第二次添加在步骤(3)中,目的是为了合成Nb3O7F纳米阵列;所述步骤(2)中,加入氢氟酸后的溶液中,氢氟酸的浓度控制在0.1~0.15mol/L,搅拌时间为5~15min,超声剥离时间为10~30min;步骤(3)中,再次加入氢氟酸的溶液中,最终氢氟酸浓度控制在0.3~0.4mol/L,搅拌时间为2~6min。
所述步骤(4)中,水热温度为120~200℃,水热时间为12~48h。
所述步骤(5)中,去离子水清洗为常温清洗,洗涤次数为3~5次,目的是除去杂质离子,提高产物纯度;乙醇清洗为常温清洗,洗涤次数为1~3次,目的是除去有机杂质;干燥温度为60~80℃,干燥时间大于5h。
所述步骤(6)中,热处理温度为400℃,热处理时间为2~4h。
有益效果:
本发明通过合理地选取原材料,充分利用原材料的物化性质,利用石墨烯表面的酸刻蚀及功能化作用,采用水热/刻蚀法在低温条件下成功制备Nb3O7F纳米阵列/石墨烯异质结材料。本发明制备过程简单,易于操作,适于放大生产。本发明所要解决的关键问题是:1)石墨烯在水溶液中的分散性和稳定性,即石墨烯在水溶液中能够长时间稳定分散,以便下一步的均匀异质成核;2)石墨烯的表面功能化,即在石墨烯表面引入氟原子,形成表面缺陷;3)含铌前驱体与石墨烯相互作用,以保证Nb3O7F在石墨烯面上异质生长;4)Nb3O7F形貌结构的控制,通过添加表面活性剂,以改变结构的微观形貌,最终获得不同形态的阵列结构。
本发明的水热/刻蚀法制备Nb3O7F纳米阵列/石墨烯异质结材料的基本原理为:制备工艺通过分散剂调节溶液的黏度,使得超声剥离的石墨烯纳米片能够在水溶液中长时间稳定分散;利用氢氟酸刻蚀石墨烯,在石墨烯面上引入氟原子,形成碳氟键的表面缺陷;通过键合作用含铌前驱体,使得Nb3O7F在面缺陷处成核生长;最后,通过非离子表面活性剂大分子的位阻效应,减缓了Nb3O7F的生长速度,并调整Nb3O7F的生长方向,最终导致不同纳米阵列在石墨烯表面的生长。
由于采用了上述方案,本发明与前述的背景技术相比,具有如下优点:
1)石墨烯的功能化方式不同,本发明是基于石墨烯的刻蚀作用,并在表面形成碳氟键,从而使石墨烯表面功能化。
2)通过低温水热的方法,首次合成Nb3O7F纳米阵列/石墨烯异质结材料,特殊的材料结构导致了良好的光电性能。
3)整个制备过程简单,操作条件易于控制,既适用于实验室研究的少量制备,又可用于大批量生产。
4)在石墨烯水溶液中添加了表面活性剂或/和络合剂,添加表面活性剂石墨烯分散性更好,结果阵列效果更好,添加络合剂可以改变阵列形貌。
附图说明
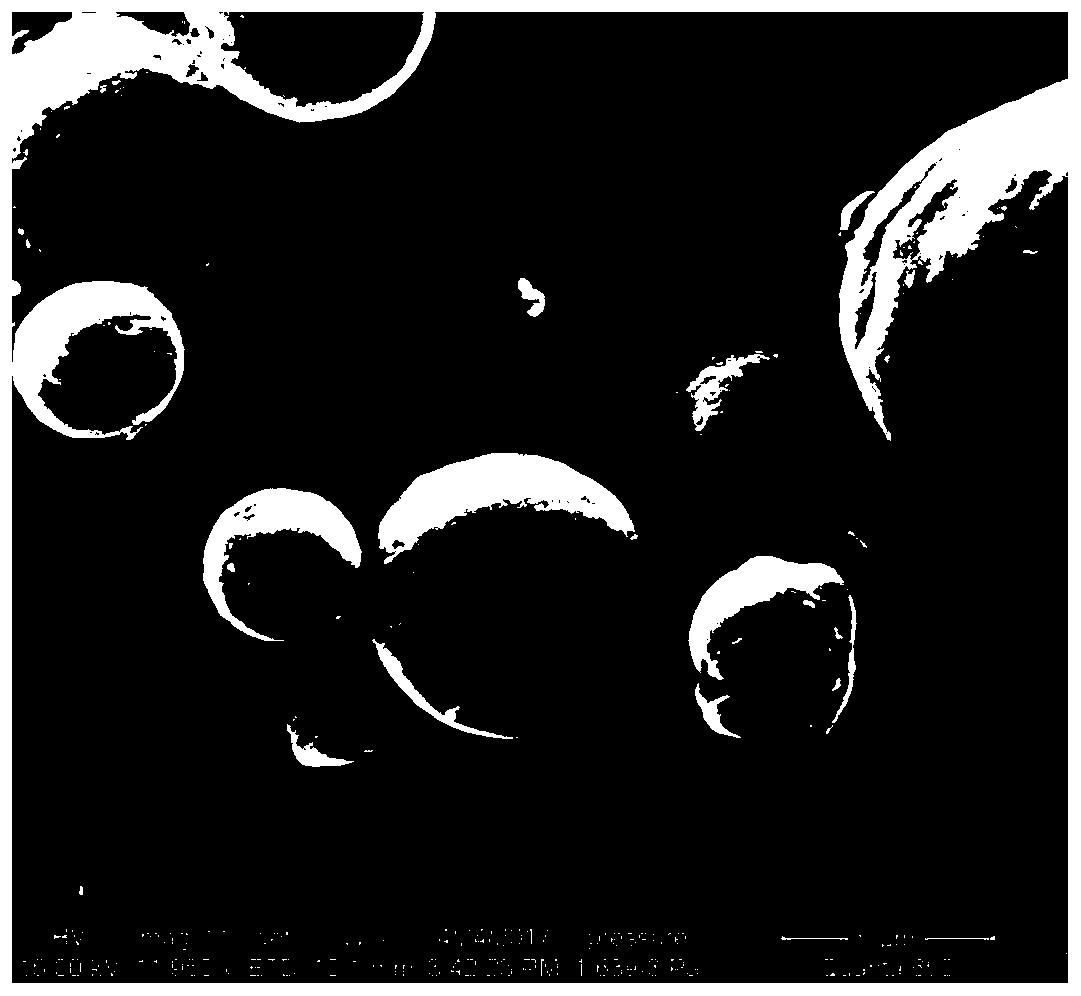
图1为本发明制备的刻蚀石墨烯的EDS图;
图2为本发明制备的刻蚀石墨烯的XPS图;
图3为本发明制备的Nb3O7F纳米柱阵列/石墨烯异质结材料的SEM图;
图4为本发明制备的Nb3O7F纳米柱阵列/石墨烯异质结材料的TEM图;
图5为本发明制备的Nb3O7F纳米柱阵列/石墨烯异质结材料的XRD图;
图6为本发明制备的Nb3O7F草坪阵列/石墨烯异质结材料的SEM图。
具体实施方式
下面结合具体实施例对本发明作更进一步的说明。
本发明采用水热/刻蚀的方法,通过一系列化学反应,在石墨烯纳米片上生长Nb3O7F 纳米阵列,步骤如下:
1)选取石墨烯纳米片作为基底,以NbCl5作为铌源,以强腐蚀性氢氟酸作为氟源,以表面活性剂和络合剂为分散剂;
2)配置石墨烯水溶液,搅拌后进行超声剥离,使石墨烯形成均匀分散液;
配置石墨烯水溶液所采用的石墨烯为纳米片结构,石墨烯的添加量,是按质量百分比,以Nb3O7F为基准,添加量为0.5~3.0wt.%;其中Nb3O7F的质量通过NbCl5换算;
搅拌时间为1~3h,超声剥离时间为4~8h
3)向上述均匀分散液中加入强腐蚀性氢氟酸,搅拌并辅以超声,使得石墨烯表面被充分刻蚀,形成碳氟键;
所述搅拌时间为5~15min,超声剥离时间为10~20min;
4)称取NbCl5粉末,加入到上述溶液中,搅拌充分后再适量添加强腐蚀性氢氟酸,继续搅拌充分;
所述搅拌时间为2~6min;
5)将上述溶液转移至特氟龙内衬的反应釜中进行水热反应;水热温度为120~200℃,水热时间为12~48h;
6)反应结束后,将产物离心分离,并用去离子水和无水乙醇清洗,在烘箱中干燥;
其中,去离子水清洗为常温清洗,洗涤次数为3~5次,除去杂质离子,提高产物纯度;乙醇清洗为常温清洗,洗涤次数为1~3次,除去有机杂质;干燥温度为60~80℃,干燥时间大于5h;
7)将干燥的产物在400℃热处理2~4h,以除去有机物,最终得到Nb3O7F纳米阵列/石墨烯异质结材料。
步骤2)中,可在配制好的石墨烯水溶液中添加表面活性剂或络合剂,或者两者都添加;
表面活性剂为三嵌段表面活性剂,表面活性剂在步骤5)的反应溶液中的浓度控制在0~40g/L。其中,三嵌段表面活性剂为P123或F127;
络合剂为一水合柠檬酸,络合剂在步骤5)的反应溶液中的浓度控制在0~40g/L。
本发明所采用的原材料包括石墨烯粉末(纯度为99.95%),NbCl5粉末(纯度为 99.99%),氢氟酸(浓度为40%),非离子表面活性剂和柠檬酸,原材料均为分析纯;
本发明中,氢氟酸采用两次添加,第一次添加在步骤(2)中,目的是为了石墨烯的表面刻蚀,从而形成碳氟键,第二次添加在步骤(3)中,目的是为了合成Nb3O7F纳米阵列;所述步骤(2)中,加入氢氟酸后的溶液中,氢氟酸的浓度控制在0.1~0.15mol/L;步骤(3)中,再次加入氢氟酸的溶液中,最终氢氟酸浓度控制在0.3~0.4mol/L。
下面结合具体实施例对本发明做进一步说明。
实施例1:
(1)称取16.9mg石墨烯加入塑料烧杯中,添加40mL H2O,配制成石墨烯水溶液,搅拌后进行超声剥离,使石墨烯形成均匀分散液;
(2)然后向上述分散液添加0.5mL HF,搅拌并辅以超声6h,使得剥离的石墨烯表面被充分刻蚀,形成碳氟键;
(3)将1.623g NbCl5加入上述溶液中,室温下磁力搅拌30min,并添加0.65mL HF 和40mL H2O,搅拌30s;
(4)将上述溶液转移至100mL聚四氟乙烯内衬中,然后将聚四氟乙烯内衬置入微型反应釜中,密封好;将反应釜放入烘箱中,在120℃下加热反应24h,然后随炉冷却;
(5)将沉淀离心分离,用常温的去离子水清洗5次,用无水乙醇清洗3次;产物在70℃的真空烘箱中干燥6h。
(6)将干燥后的产物在400℃的空气中热处理2h,即得到最终产物。
实施例2:
(1)称取16.9mg石墨烯加入烧杯中,添加40mL H2O,配制成石墨烯水溶液,然后添加2g P123,在70℃下水浴搅拌30min,使P123完全溶解;然后进行超声剥离,使石墨烯形成均匀分散液;
(2)然后向上述分散液添加0.5mL HF,搅拌并辅以超声6h,使得剥离的石墨烯表面被充分刻蚀,形成碳氟键;
(3)称取1.623g NbCl5加入上述溶液中,室温下磁力搅拌1h;并添加0.65mL HF 和40mL H2O,搅拌30s;
(4)将搅拌后的混合溶液转移至100mL聚四氟乙烯内衬中,然后将聚四氟乙烯内衬置入微型反应釜中,密封好;将反应釜转移至烘箱中,在120℃下加热反应24h,然后随炉冷却;
(5)将沉淀离心分离,用去离子水清洗5次,用无水乙醇清洗3次;将产物在70℃的真空烘箱中干燥6h;
(6)最后,将干燥后的产物在400℃的空气中进行热处理3h,即得最终产物。
实施例3:
(1)称取16.9mg石墨烯加入烧杯中,添加40mL H2O,配制成石墨烯水溶液,然后添加2g柠檬酸,在70℃下水浴搅拌30min,使柠檬酸完全溶解;然后进行超声剥离,使石墨烯形成均匀分散液;
(2)然后向上述分散液添加0.5mL HF,搅拌并辅以超声5h,使得剥离的石墨烯表面被充分刻蚀,形成碳氟键;
(3)称取1.623g NbCl5加入上述溶液中,室温下磁力搅拌1h;并添加0.65mL HF 和40mL H2O,搅拌30s;
(4)将搅拌后的混合溶液转移至100mL聚四氟乙烯内衬中,然后将聚四氟乙烯内衬置入微型反应釜中,密封好;将反应釜转移至烘箱中,在120℃下加热反应24h,然后随炉冷却;
(5)将沉淀离心分离,用去离子水清洗5次,用无水乙醇清洗3次;将产物在70℃的真空烘箱中干燥6h;
(6)最后,将干燥后的产物在400℃的空气中进行热处理3h,即得最终产物。
实施例4:
(1)称取16.9mg石墨烯加入烧杯中,添加40mL H2O,配制成石墨烯水溶液,然后添加2g F127和2g柠檬酸,在70℃下水浴搅拌30min,使柠檬酸完全溶解;然后进行超声剥离,使石墨烯形成均匀分散液;
(2)然后向上述分散液添加0.5mL HF,搅拌并辅以超声5h,使得剥离的石墨烯表面被充分刻蚀,形成碳氟键;
(3)称取1.623g NbCl5加入上述溶液中,室温下磁力搅拌1h;并添加0.65mL HF 和40mL H2O,搅拌30s;
(4)将搅拌后的混合溶液转移至100mL聚四氟乙烯内衬中,然后将聚四氟乙烯内衬置入微型反应釜中,密封好;将反应釜转移至烘箱中,在120℃下加热反应24h,然后随炉冷却;
(5)将沉淀离心分离,用去离子水清洗5次,用无水乙醇清洗3次;将产物在70℃的真空烘箱中干燥6h;
(6)最后,将干燥后的产物在400℃的空气中进行热处理4h,即得最终产物。
图1为本发明制备的刻蚀石墨烯的EDS图,从图中可以知道,刻蚀后的石墨烯含有碳元素和氟元素,且氟元素在石墨烯上均匀分布。
图2为本发明制备的刻蚀石墨烯的XPS图,从图中可以知道,刻蚀前后的石墨烯最大的变化就是刻蚀后石墨烯表面形成了碳氟键。
图3为本发明制备的Nb3O7F纳米柱阵列/石墨烯异质结材料的SEM图,从图中可以看出产物为纳米柱阵列。
图4为本发明制备的Nb3O7F纳米柱阵列/石墨烯异质结材料的TEM图,从图中可以看出超声后纳米柱阵破碎,纳米柱直径在20~50nm范围内。
图5为本发明制备的Nb3O7F纳米柱阵列/石墨烯异质结材料的XRD图,从图中可以看出产物的主要物相为Nb3O7F(JCPDS No.18-0915),这主要源自于石墨烯含量比较低的缘故。
图6为本发明制备的Nb3O7F草坪阵列/石墨烯异质结材料的SEM图,从图中可以看出产物为草坪阵列。
上述列举出了本发明的四种实施方式,但本发明的上述实施方案都只是对本发明的说明而不能限制本发明,权利要求书指出本发明的范围。因此,在不违背本发明基本思想的情况下,只要利用HF对石墨烯进行表面刻蚀,形成碳氟键,并利用这种表面缺陷形成Nb3O7F/石墨烯异质结材料都应是落入本发明的保护范围内。本发明各原材料的上下限、区间取值,以及工艺参数(如温度、时间等)的上下限、区间取值都能实现本发明,在此不一一列举。
一种NbOF纳米阵列/石墨烯异质结复合材料的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0