








专利摘要
本发明提供了具有式I所示结构的三萜衍生物,属于有机合成技术领域。本发明提供的具有式I所示结构的三萜衍生物具有明确的选择性抗艾滋病毒活性。
权利要求
1.具有式I所示结构的三萜衍生物,
2.权利要求1所述的三萜衍生物的制备方法,其特征在于,包括以下步骤:
将三萜皂苷类化合物beesioside I、水解酶molsin和磷酸氢二钠-柠檬酸缓冲液混合进行反应,得到Beesioside I的苷元;
将所述Beesioside I的苷元分别与2,2-二甲基琥珀酸酐、2,2-二甲基戊二酸酐和二甘醇酐混合进行微波反应,得到具有式I所示结构的三萜衍生物。
3.根据权利要求2所述的制备方法,其特征在于,所述三萜皂苷类化合物beesioside I与水解酶molsin的质量比为1:1~1:10。
4.根据权利要求2或3所述的制备方法,其特征在于,所述磷酸氢二钠-柠檬酸缓冲液的pH值为4.0。
5.根据权利要求2所述的制备方法,其特征在于,所述微波反应的温度为150~160℃,所述微波反应的时间为1~ 3 h。
6.权利要求1所述的具有式I所示结构的三萜衍生物或权利要求2~5任一项所述制备方法制得的具有式I所示结构的三萜衍生物在制备抗艾滋病药物中的应用。
7.根据权利要求6所述的应用,其特征在于,所述抗艾滋病药物包含有效剂量的具有式I所示结构的三萜衍生物、其立体异构体、可药用盐和药学上可接受的载体、辅料、赋形剂和稀释剂。
8.根据权利要求6或7所述的应用,其特征在于,所述抗艾滋病药物的剂型包括片剂、注射剂、胶囊剂、颗粒剂、丸剂、散剂、口服液、缓释制剂、控释制剂或纳米制剂药学上可接受的剂型。
9.根据权利要求6或7所述的应用,其特征在于,当所述R为1基团时,所述具有式I所示结构的三萜衍生物具有显著的体外抗HIV-1作用,在HIV-1
说明书
技术领域
本发明涉及有机合成技术领域,尤其涉及具有式I所示结构的三萜衍生物及其制备方法和应用。
背景技术
艾滋病(Acquired Immunodeficiency Syndrome,AIDS),又称获得性免疫缺陷综合征,是由于感染艾滋病毒(Human Immunodeficiency Virus,HIV) 而引起的一种严重威胁人类健康、影响社会发展的重大疾病。目前仍然没有研发出能够治愈艾滋病的药物,使得大量的患者死于并发症;同时,这一公共卫生问题增加了国家财政的压力,延缓了经济的发展。因此,艾滋病的预防与治疗问题亟待解决。
迄今,还未找到能够彻底治愈HIV感染的理想药物,而对于艾滋病的治疗主要是在于延缓病程和消除并发症。高效抗逆转录病毒治疗(HAART),又称“鸡尾酒疗法”,为一种联合用药方法,即两种或两种以上逆转录酶抑制剂与一种或一种以上蛋白酶抑制剂联合应用,从而抑制HIV在人体内的复制。而HAART组合的药物在临床应用中出现了诸多的毒副作用:核苷类逆转录酶抑制剂能够通过阻断DNA聚合酶而诱导线粒体毒性,可能引起神经肌肉毒性、胰腺炎、高乳酸血症、贫血和中性粒细胞减少等严重的副作用;非核苷类逆转录酶抑制剂具有肝毒性,可造成肝损伤,且在使用过程中可能发生严重的皮疹以及中枢神经毒性的不良反应;蛋白酶抑制剂长期应用将会导致脂代谢紊乱,继而诱发动脉粥样硬化、心肌梗死等心脑血管疾病。在联合用药过程中,这些药物相互作用而引起的不良反应可能更加突出,并且因为长期用药,耐药性的发生率较高,往往需要一段时间后更换新的药物组合继续治疗。
我国传统中医药理论体系历史悠久,人们在长期的临床实践中,对各类中药的疗效、毒性等方面积累了丰富的经验。通过现代科学技术手段,中药作用的物质基础逐渐被挖掘出来,再运用有机合成,对先导化合物的结构进行修饰,以提高有效成分的活性,降低毒副作用,改善其吸收、分布、代谢与排泄,提高稳定性,进而获得可用于生产的单体药物。据文献报道,天然环菠萝蜜烷三萜类化合物黄芪甲苷和环黄芪醇能够通过调节上端粒酶的活性促使HIV患者的CD8+T淋巴细胞增殖(Yung LY,Lam WS,Ho MK,et al. Astragaloside IVand cycloastragenol stimulate the phosphorylation of extracellular signal-regulated protein kinase in multiple cell types.Planta Med, 2012,78(2):115-121.);从五味子科植物中也发现了一系列可抗HIV活性的环菠萝蜜烷三萜类化合物[金银萍,焉石,刘俊霞,等.五味子科植物中环阿屯烷型三萜类成分及其药理作用研究进展.中草药,2014,45(4):582-589]。
发明内容
鉴于此,本发明的目的在于提供具有式I所示结构的三萜衍生物及其制备方法和应用。本发明提供的具有式I所示结构的三萜衍生物具有明确的选择性抗艾滋病毒活性。
为了实现上述发明目的,本发明提供以下技术方案:
具有式I所示结构的三萜衍生物。
本发明还提供了上述技术方案所述的三萜衍生物的制备方法,包括以下步骤:
将三萜皂苷类化合物beesioside I、水解酶molsin和磷酸氢二钠-柠檬酸缓冲液混合进行反应,得到Beesioside I的苷元;
将所述Beesioside I的苷元分别与2,2-二甲基琥珀酸酐、2,2-二甲基戊二酸酐和二甘醇酐混合进行微波反应,得到具有式I所示结构的三萜衍生物。
优选地,所述三萜皂苷类化合物beesioside I与水解酶molsin的质量比为1:1~1:10。
优选地,所述磷酸氢二钠-柠檬酸缓冲液的pH值为4.0。
优选地,所述微波反应的温度为150~160℃,所述微波反应的时间为1~3h。
本发明还提供了上述技术方案所述的具有式I所示结构的三萜衍生物在制备抗艾滋病药物中的应用。
优选地,所述抗艾滋病药物包含有效剂量的具有式I所示结构的三萜衍生物、其衍生物、其立体异构体、可药用盐和药学上可接受的载体、辅料、赋形剂和稀释剂。
优选地,所述抗艾滋病药物的剂型包括片剂、注射剂、胶囊剂、颗粒剂、丸剂、散剂、口服液、缓释制剂、控释制剂或纳米制剂药学上可接受的剂型。
优选地,当所述R为1基团时,所述具有式I所示结构的三萜衍生物具有显著的体外抗HIV-1作用,在HIV-1NL4-3病毒感染的MT-4细胞中,对HIV 病毒具有明显的抑制作用,半数有效抑制浓度EC50为0.025μM,而治疗指数TI大于800。
本发明提供了具有式I所示结构的三萜衍生物,具有明确的选择性抗艾滋病毒活性。
附图说明
图1为本发明具有式I所示结构的三萜衍生物的制备方法的原理图。
具体实施方式
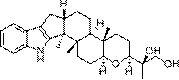
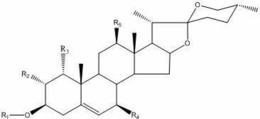
本发明提供了具有式I所示结构的三萜衍生物,
在本发明中,当R为1时,所述具有式I所示结构的三萜衍生物的化学名称为:(20S,24S)-15β,16β-diacetoxy-18,24,20,24-diepoxy-9,19-cyclolanostane -3β,25-diol 3-O-3′,3′-dimethylsuccinate,当R为2时,所述具有式I所示结构的三萜衍生物的化学名称为:(20S,24S)-15β,16β-diacetoxy-18,24;20,24- diepoxy-9,19-cyclolanostane-3β,25-diol 3-O-4′,4′-dimethylglutarate,当R为3 时,所述具有式I所示结构的三萜衍生物的化学名称为:(20S,24S)-15β,16β-diacetoxy-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,25- diol 3-O-diglycolate。
本发明还提供了上述技术方案所述的三萜衍生物的制备方法,包括以下步骤:
将三萜皂苷类化合物beesioside I、水解酶molsin和磷酸氢二钠-柠檬酸缓冲液混合进行反应,得到Beesioside I的苷元;
将所述Beesioside I的苷元、分别与2,2-二甲基琥珀酸酐、2,2-二甲基戊二酸酐和二甘醇酐混合进行微波反应,得到具有式I所示结构的三萜衍生物。
本发明将三萜皂苷类化合物beesioside I、水解酶molsin和磷酸氢二钠- 柠檬酸缓冲液混合进行反应,得到Beesioside I的苷元。
在本发明中,所述三萜皂苷类化合物beesioside I具有式II所示的结构:
本发明对所述三萜皂苷类化合物beesioside I的来源没有特殊的限定,采用本领域技术人员熟知的制备方法制得即可。在本发明的实施例中,所述三萜皂苷类化合物beesioside I优选通过以下步骤得到:
取黄三七Souliea vaginata(Maxim.)Franch.10Kg,干燥粉碎后,采用不同倍量、不同比例乙醇/水、甲醇/水或者丙酮/水回流或冷浸提取,减压回收溶剂得提取物220g,提取物溶解于水里,依次经过石油醚、三氯甲烷、乙酸乙酯和正丁醇萃取,乙酸乙酯萃取部位35g经硅胶(100~200目)柱层析,石油醚/乙酸乙酯(100:0~1:1)、二氯甲烷/甲醇(50:1~1:1)洗脱,获得极性不同的流份,取中等极性的流份8g,经硅胶柱层析(200~300目)石油醚/乙酸乙酯(10:1~1:1)、二氯甲烷/甲醇(20:1~5:1)洗脱,并经反相柱层析甲醇/水(50:50~100:0)洗脱,并经凝胶LH-20甲醇洗脱得活性前体化合物粗品,最后经甲醇重结晶得到三萜皂苷类化合物beesioside I。
在本发明中,所述三萜皂苷类化合物beesioside I与水解酶molsin的质量比优选为1:1~1:10。
在本发明中,所述磷酸氢二钠-柠檬酸缓冲液的pH值优选为4.0。
在本发明中,所述混合优选在无水乙醇中进行。在本发明中,所述混合优选先将三萜皂苷类化合物beesioside I用无水乙醇溶解,向该溶液中加入溶解于纯水的水解酶molsin(Aspergillus saitoi)和0.2M磷酸氢二钠-0.1M柠檬酸缓冲液(pH 4.0)。
在本发明中,所述反应的温度优选为37℃,所述反应的时间优选为2 天。
反应结束后,本发明优选将所得反应液用等体积的乙酸乙酯萃取3次,乙酸乙酯部分合并,经无水硫酸钠干燥、浓缩后,经硅胶柱(200~300目) 层析,用正己烷/丙酮(10:1~1:1)洗脱,并经甲醇重结晶,得到Beesioside I的苷元。
在本发明中,所述Beesioside I的苷元的结构如式III所示:
得到Beesioside I的苷元后,本发明将所述Beesioside I的苷元分别与2,2- 二甲基琥珀酸酐、2,2-二甲基戊二酸酐和二甘醇酐混合进行微波反应,得到具有式I所示结构的三萜衍生物。
当所述Beesioside I的苷元与2,2-二甲基琥珀酸酐进行微波反应时,得到 R为1的基团的具有式I所示结构的三萜衍生物;当所述Beesioside I的苷元与2,2-二甲基戊二酸酐进行微波反应时,得到R为2的基团的具有式I所示结构的三萜衍生物;当所述Beesioside I的苷元与二甘醇酐进行微波反应时,得到R为3的基团的具有式I所示结构的三萜衍生物。
在本发明中,所述微波反应优选在无水吡啶和4-二甲氨基吡啶(DMAP) 中进行。
在本发明中,所述Beesioside I的苷元与2,2-二甲基琥珀酸酐、2,2-二甲基戊二酸酐和二甘醇酐的摩尔比优选均为1:1~1:10。
在本发明中,所述微波反应的温度优选为150~160℃,所述微波反应的时间优选为1~3h。
微波反应完成后。本发明优选将反应液加入1N盐酸中和,然后加入乙酸乙酯萃取3次,乙酸乙酯部分用brine洗涤3次,加入无水硫酸镁干燥,经硅胶柱(200~300目)层析,正己烷/丙酮梯度洗脱(10:1~1:1),得到具有式I所示结构的三萜衍生物。
本发明还提供了上述技术方案所述的具有式I所示结构的三萜衍生物在制备抗艾滋病药物中的应用。
在本发明中,所述抗艾滋病药物优选包含有效剂量的具有式I所示结构的三萜衍生物、其衍生物、其立体异构体、可药用盐和药学上可接受的载体、辅料、赋形剂和稀释剂。
在本发明中,所述抗艾滋病药物的剂型优选包括片剂、注射剂、胶囊剂、颗粒剂、丸剂、散剂、口服液、缓释制剂、控释制剂或纳米制剂药学上可接受的剂型。
在本发明中,当所述R为1基团时,所述具有式I所示结构的三萜衍生物具有显著的体外抗HIV-1作用,在HIV-1NL4-3病毒感染的MT-4细胞中,对HIV病毒具有明显的抑制作用,半数有效抑制浓度EC50为0.025μM,而治疗指数TI大于800。
下面结合实施例对本发明提供的具有式I所示结构的三萜衍生物及其制备方法和应用进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
图1为本发明具有式I所示结构的三萜衍生物的制备方法的原理图。
实施例1
三萜皂苷类化合物beesioside I的制备
取黄三七Souliea vaginata(Maxim.)Franch.药材10Kg,干燥粉碎后,采用不同倍量、不同比例乙醇/水、甲醇/水或者丙酮/水回流或冷浸提取,减压回收溶剂得提取物220g,提取物溶解于水里,依次经过石油醚、三氯甲烷、乙酸乙酯和正丁醇萃取,乙酸乙酯萃取部位35g经硅胶(100~200目,200g) 柱层析,石油醚/乙酸乙酯(100:0~1:1)、二氯甲烷/甲醇(50:1~1:1) 洗脱,获得极性不同的流份,取中等极性的流份8g,经硅胶柱层析(200~300目,150g)石油醚/乙酸乙酯(10:1~1:1)、二氯甲烷/甲醇(20:1~5:1) 洗脱,并经反相柱层析甲醇/水(50:50~100:0)洗脱,并经凝胶LH-20 甲醇洗脱得活性前体化合物粗品,最后经甲醇重结晶得单体化合物1.1g,经核磁共振波谱和质谱,并与参考文献(N.Sakurai,M.Nagai,T.Goto,T.Inoue, P.G.Xiao,Studies on the constituents ofBeesiacalthaefolia and Souliea vaginata. IV.1)Beesioside I,a cyclolanostanolxyloside from the rhizomes of Beesia calthaefolia,Chem.Pharm.Bull.,1993,41,272-275)比对,确定为三萜皂苷类化合物beesioside I。
Beesioside I,白色粉末,ESI-MS:m/z 743[M+Na]
实施例2
1)制备beesioside I的苷元
将beesioside I(1.1g,1.53mmol)用100mL无水乙醇溶解,向该溶液中加入溶解于100mL纯水的水解酶molsin(Aspergillus saitoi)2.2g和0.2M 磷酸氢二钠-0.1M柠檬酸缓冲液(pH 4.0)1000mL,此溶液体系在37℃下搅拌反应2天。反应液用等体积的乙酸乙酯萃取3次,乙酸乙酯部分合并,经无水硫酸钠干燥、浓缩后,经硅胶柱(200~300目)层析,用正己烷/丙酮(10: 1~1:1)洗脱,并经甲醇重结晶,得到单体化合物,经核磁共振波谱和质谱,并与参考文献(N.Sakurai,M.Nagai,T.Goto,T.Inoue,P.G.Xiao,Studies on theconstituents ofBeesia calthaefolia and Souliea vaginata.IV.1)Beesioside I,acyclolanostanol xyloside from the rhizomes ofBeesia calthaefolia,Chem.Pharm.Bull.,1993,41,272-275)比对,确定为Beesioside I的苷元,无色晶体;ESIMS m/z 589[M+H]
2)制备化合物1~3
在10mL无水吡啶中,加入上述步骤得到的beesioside I苷元(0.06 mmol),等当量的4-dimethylaminopyridine(DMAP),分别加入10倍当量的2,2-二甲基琥珀酸酐、2,2-二甲基戊二酸酐和二甘醇酐,155℃微波反应2h,反应停止后,反应液加入1mL 1N盐酸中和,然后加入10mL乙酸乙酯萃取 3次,乙酸乙酯部位用brine洗涤3次,加入无水硫酸镁干燥,经硅胶柱 (200~300目)层析,正己烷/丙酮梯度洗脱(10:1~1:1),得到 (20S,24S)-15β,16β-diacetoxy-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,25-di ol 3-O-3′,3′-dimethylsuccinate(1,得率37.2%),(20S,24S)-15β,16β -diacetoxy-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,25-diol 3-O-4′,4′ -dimethylglutarate(2,得率35.7%)和(20S,24S)-15β,16β-diacetoxy-18,24;20,24 -diepoxy-9,19-cyclolanostane-3β,25-diol 3-O-diglycolate(3,得率42.6%)。
3)化合物的结构确定
化合物1,无色晶体,熔点221-223℃(MeOH);[α]20D–12.0(c 0.10, MeOH);ESIMS:m/z 717[M+H]
化合物2,无色晶体,m.p.228-230℃(MeOH);[α]20D–13.6(c 0.15, MeOH);ESIMS:m/z 731[M+H]
化合物3,无色晶体,m.p.197-199℃(MeOH);[α]20D–1.9(c 0.16, MeOH);ESIMS:m/z 727[M+Na]
实施例3
式I化合物在HIV-1NL4-3感染的MT-4细胞中抑制HIV活性试验
待测试活性的药物:
(20S,24S)-15β,16β-diacetoxy-18,24;20,24-diepoxy-9,19-cyclolanostane-3β, 25-diol 3-O-3′,3′-dimethylsuccinate(1)
(20S,24S)-15β,16β-diacetoxy-18,24;20,24-diepoxy-9,19-cyclolanostane-3β, 25-diol 3-O-4′,4′-dimethylglutarate(2)
(20S,24S)-15β,16β-diacetoxy-18,24;20,24-diepoxy-9,19-cyclolanostane-3β, 25-diol 3-O-diglycolate(3)
测试方法:
样品对HIV-1抑制作用的体外评价试验参照文献进行Z.Dang,L.Zhu,W. Lai,H.Bogerd,K.H.Lee,L.Huang,C.H.Chen,Aloperine and its derivatives as a newclass ofHIV-1entry inhibitors.ACS Med.Chem.Lett.7(2016)240-244. HIV-1NL4-3Nanoluc-sec病毒感染的MT4细胞培养于9孔板中,加入不同浓度的待测化合物,HIV-1NL4-3Nanoluc-sec病毒是一种报告病毒,具有 secNluc作为报告基因,化合物用DMSO溶解后,采用Promega Nano-Glo LuciferaseAssay System通过检测荧光素激酶的活性来检测病毒的复制。测定结果见表1。由表1可以看出,(20S,24S)-15β,16β-diacetoxy-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,25-diol 3-O-3′,3′-dimethylsuccinate(1)被认为是最有效的抗HIV物质,其半数有效抑制HIV浓度EC50值为0.025μM,且治疗指数TI值大于800,与首个进入II期临床试验的HIV成熟期抑制剂 3-O-(3′,3′-dimethylsuccinyl)-betulinic acid(DSB)相差无几,有可能发展成为天然来源的抗HIV-1药物。
表1样品及阳性药物在MT-4细胞上抑制HIV-1活性试验结果
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
具有式I所示结构的三萜衍生物及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。








动态评分
0.0