

IPC分类号 : C07F19/00,C07F5/00,C07F7/08,C08F136/08,C08F4/52,C09K11/06






专利摘要
本发明涉及一种双吡咯甲烯型稀土金属配合物、制备方法及应用,属于催化剂技术领域。所述配合物催化活性和选择性较高,还具有荧光性,结构如结构式Ⅰ和Ⅱ所示:先制备双吡咯甲烯型配体,再制备所述配合物,原料价廉易得且易修饰,经济效率高,环保性好,适合工业化生产。所述配合物、烷基铝试剂和有机硼盐组成的催化体系,可催化均聚或共聚反应,得到具有荧光性质的聚合材料;聚合时双吡咯甲烯基配体与烷基铝的置换反应证明配位聚合机理的存在;所述配合物在催化异戊二烯的聚合反应中表现出高活性,顺‑1,4‑聚合选择性高达97%,获得具有荧光性质的聚异戊二烯。
权利要求
1.一种双吡咯甲烯型稀土金属配合物,其特征在于:所述稀土金属配合物的结构如结构式Ⅰ和结构式Ⅱ所示:
R
R
R
Ln是稀土金属。
2.一种如权利要求1所述的双吡咯甲烯型稀土金属配合物的制备方法,其特征在于:步骤如下:
(1)制备双吡咯甲烯型配体
在保护气体氛围下将芳香醛和取代的吡咯加入到反应器中,加入二氯甲烷作溶剂,滴加加入三氟乙酸,反应2h~4h后,加入DDQ进行氧化脱氢反应15min~30min,得到产物,将产物萃取、干燥以及过滤,将滤液用柱层析纯化,得到双吡咯甲烯型配体;
保护气体为氮气或惰性气体;
取代的吡咯中的取代基为所述R
芳香醛、取代的吡咯和DDQ的摩尔比为1:2:1.05~1.2,三氟乙酸摩尔量为芳香醛摩尔量的0.1%~0.5%;
(2)制备双吡咯甲烯型稀土金属配合物
将所述双吡咯甲烯型配体加入到反应器中,用甲苯作溶剂,得到混合物h;将混合物h滴加到溶解有金属源的甲苯溶液中,在20℃~25℃搅拌反应2h~6h,得到反应液;除去反应液中的溶剂得到固体,用正己烷洗涤3次~5次后,除去溶剂,得到的固体粉末用正己烷和四氢呋喃的混合溶剂或正己烷和甲苯的混合溶剂溶解,在-20℃~-35℃放置结晶,得到晶体为所述双吡咯甲烯型稀土金属配合物;
步骤(2)在无水无氧环境下进行;金属源为含有Ln的化合物。
3.根据权利要求2所述的一种双吡咯甲烯型稀土金属配合物的制备方法,其特征在于:步骤(1)中:将DDQ用二氯甲烷溶解后滴加加入;柱层析使用的洗脱剂为二氯甲烷和甲醇的混合溶液,二氯甲烷与甲醇的体积比为99:1;
步骤(2)中:无水无氧环境采用在手套箱中实现;金属源为二四氢呋喃-三(三甲基硅亚甲基)稀土金属化合物;双吡咯甲烯型配体与金属源的摩尔比为1:1;将混合物h置于手套箱中,冷冻后再滴加到溶解有金属源的甲苯溶液中。
4.一种如权利要求1所述的双吡咯甲烯型稀土金属配合物的应用,其特征在于:将所述稀土金属配合物作为催化剂和荧光探针,与烷基铝试剂和有机硼盐一起用于:
(1)α-烯烃、环烯烃、共轭二烯烃或非共轭二烯烃的均聚反应;或
(2)α-烯烃、环烯烃、共轭二烯烃和非共轭二烯烃中任意两种的共聚反应;或
(3)α-烯烃、环烯烃、共轭二烯烃和非共轭二烯烃中的任一种与二氧化碳的共聚反应;
有机硼盐与所述稀土金属配合物的摩尔比为1~2:1;
烷基铝试剂与所述稀土金属配合物的摩尔比为0.5~50:1;
α-烯烃、环烯烃、共轭二烯烃和非共轭二烯烃中任一种与所述稀土金属配合物的摩尔比为200~5000:1。
5.根据权利要求4所述的一种双吡咯甲烯型稀土金属配合物的应用,其特征在于:所述均聚反应步骤如下:
(1)向反应器中依次加入良溶剂、烷基铝试剂、所述稀土金属配合物、单体a和有机硼盐,-30℃~120℃搅拌反应0.1h~72h,得到置换后的稀土金属烷基配合物和铝配合物,以及聚合后的反应液;
步骤(1)在无水无氧条件下进行;
(2)向反应器中加入链终止剂,使反应中止,得到终止聚合反应的聚合物反应液;将聚合物反应液用无水乙醇进行沉降,析出固体物质,将固体物质在40℃~60℃真空干燥,除去溶剂至恒重,得到均聚产物;
烷基铝试剂、单体a、有机硼盐与所述稀土金属配合物的摩尔比为0.5~50:200~5000:1~2:1;
单体a为α-烯烃、环烯烃、共轭二烯烃或非共轭二烯烃;
所述共聚反应步骤如下:
(1)向反应器中依次加入良溶剂、烷基铝试剂、所述稀土金属配合物、单体b和有机硼盐,-60℃~120℃搅拌反应0.1h~72h,得到置换后的稀土金属烷基配合物和铝配合物,以及聚合后的反应液;
步骤(1)在无水无氧条件下进行;
(2)向反应器中加入链终止剂,使反应中止,得到终止聚合反应的聚合物反应液;将聚合物反应液用无水乙醇进行沉降,析出固体物质,将固体物质在40℃~60℃真空干燥,除去溶剂至恒重,得到共聚产物;
烷基铝试剂、单体b、有机硼盐和所述稀土金属配合物的摩尔比为0.5~50:200~5000:1~2:1;
单体b为α-烯烃、环烯烃、共轭二烯烃和非共轭二烯烃中的任两种;或
单体b为α-烯烃、环烯烃、共轭二烯烃和非共轭二烯烃中的任一种与二氧化碳。
6.根据权利要求5所述的一种双吡咯甲烯型稀土金属配合物的应用,其特征在于:良溶剂为正己烷、正庚烷、苯、甲苯、环己烷、氯苯、邻二氯苯、间二氯苯、对二氯苯、三氯苯和四氢呋喃中的一种以上;
链终止剂为含有2,6-二叔丁基-4-甲基苯酚、2,3,4-三甲基苯酚、间二苯酚、2,6-二乙基苯酚或对叔丁基苯酚的乙醇溶液。
7.根据权利要求6所述的一种双吡咯甲烯型稀土金属配合物的应用,其特征在于:步骤(1)的无水无氧条件采用手套箱实现;
2,6-二叔丁基-4-甲基苯酚、2,3,4-三甲基苯酚、间二苯酚、2,6-二乙基苯酚或对叔丁基苯酚的质量分数为5%~15%。
8.根据权利要求4~7任一项所述的一种双吡咯甲烯型稀土金属配合物的应用,其特征在于:烷基铝试剂是分子式为AlX
有机硼盐为三苯基(甲基)-四(五氟苯)硼盐、苯基-二甲基氨基-四(五氟苯)硼盐、苯基-二甲基氨基-四苯基硼盐或三(五氟苯)硼盐。
9.根据权利要求8所述的一种双吡咯甲烯型稀土金属配合物的应用,其特征在于:烷基铝为三甲基铝、三乙基铝、三正丙基铝、三正丁基铝、三异丙基铝、三异丁基铝、三己基铝、三环己基铝或三辛基铝;
烷基氢化铝为氢化二甲基铝、氢化二乙基铝、氢化二正丙基铝、氢化二正丁基铝、氢化二异丙基铝、氢化二异丁基铝、氢化二戊基铝、氢化二己基铝、氢化二环己基铝或氢化二辛基铝;
烷基氯化铝为氯化二甲基铝、氯化二乙基铝、氯化二正丙基铝、氯化二正丁基铝、氯化二异丙基铝、氯化二异丁基铝、氯化二戊基铝、氯化二己基铝、氯化二环己基铝或氯化二辛基铝;
铝氧烷为甲基铝氧烷、乙基铝氧烷、正丙基铝氧烷或正丁基铝氧烷;
α-烯烃为乙烯、丙烯、1-丁烯、1-戊烯、4-甲基-1-戊烯、1-己烯、1-庚烯、1-辛烯、1-癸烯、1-十二碳烯、1-十四碳烯、1-十六碳烯、1-二十碳烯、苯乙烯、α-甲基苯乙烯或3-氯甲基苯乙烯;
环烯烃为降冰片烯、极性降冰片烯、降冰片二烯、亚乙基降冰片烯、苯基降冰片烯、乙烯基降冰片烯或双环戊二烯;
共轭二烯烃为1,3-丁二烯、异戊二烯、月桂烯、1,3-环己二烯、罗勒烯或环戊二烯;
非共轭二烯烃为间戊二烯、1,5-己二烯、1,6-庚二烯或1,7-辛二烯。
10.根据权利要求9所述的一种双吡咯甲烯型稀土金属配合物的应用,其特征在于:单体a为异戊二烯。
说明书
技术领域
本发明涉及一种双吡咯甲烯型稀土金属配合物、制备方法及应用,属于稀土金属材料技术领域。
背景技术
非茂配体与稀土金属配位合成稀土金属催化剂,一直以来都是非茂配体研究的主要方向。由于稀土金属中的镧系元素含有4f轨道,多数稀土金属在其化合物中一般是以+3价氧化态稳定存在的,因此稀土金属有机配合物具有其既有别于d族过渡金属有机配合物,又有别于主族金属有机配合物的独特性质。稀土金属离子半径大、配位数高,更有利于底物的配位和活化。4f轨道由于受到极强的屏蔽作用不参与成键,d族的18电子规则在稀土金属有机化学中不适用,也不易发生d族过渡金属有机配合物化学中常见的氧化加成和还原消除反应。稀土元素虽属于副族元素,但稀土-碳σ键、稀土-氮σ键却具有较强的离子性,有好的反应活性。稀土离子属于硬Lewis酸,易于和含N、O原子的硬碱配体配位,表现出强的亲氧性,而与有机膦、烯烃及一氧化碳等软碱配位作用弱。稀土元素具有亲氧性、强还原性,Lewis酸性和高配位数的特点,能进行轨道对称性不允许的反应。因此,稀土金属有机化合物有很多重要的化学性质和物理性能,在催化有机合成和高分子合成(如烯烃配位聚合)中表现出很多独特的性能。
非茂金属催化剂相对于传统的Ziegler-Natta催化体系和茂金属催化体系相比,与茂金属催化剂一样,属于均相催化体系,而且是单一活性中心,利于高分子配位聚合机理的研究,活性和选择性都可以随着配体选择的不同来控制,除此之外,还拥有与茂金属催化剂不同的优势:非茂金属催化体系配体的选择性范围广,合成简单,而且利于修饰,在国外的专利覆盖面小,可以有很大突破。在非茂体系中,一系列杂原子基团被巧妙地运用,配体中的胺基、胍基、脒基、烷氧基、吡咯、吡唑和碳硼烷等的运用,可合成极具潜力的一系列配体。因此,非茂金属催化剂也成为近年来金属有机化合物和高分子配位化学领域的新热点。
由于双吡咯甲烯配体具有独特的刚性共轭平面,在与过渡金属配位后,表现出了良好的光学特性而被广泛的研究,除此之外,双吡咯甲烯配体的各个位点容易修饰,根据需要引入不同位阻大小,不同供电子能力的官能团,同时,双吡咯甲烯配体中-C=N-键的存在提供了孤电子,使得双吡咯甲烯配体具有极大的灵活性和良好的配位能力,但是双吡咯甲烯-金属配合物作为催化剂的研究还没有被广泛的研究,应用于高分子聚合中的报道少之又少。
发明内容
为克服现有技术存在的缺陷,本发明的目的之一在于提供一种双吡咯甲烯型稀土金属配合物,所述稀土金属配合物具有较高的催化活性和选择性,还具有荧光性。
本发明的目的之二在于提供一种双吡咯甲烯型稀土金属配合物的制备方法,所述方法经济效率高,环保性好,适合工业化生产。
本发明的目的之三在于提供一种双吡咯甲烯型稀土金属配合物的应用,所述应用为催化剂和荧光探针。
具体地说,所述稀土金属配合物、烷基铝试剂和有机硼盐组成的反应体系,可催化完成直链烯烃、支链烯烃以及极性单体等的均聚及共聚反应,得到一系列具有特定结构的聚合材料;特别是当单体a为异戊二烯时,所述稀土金属配合物在催化异戊二烯聚合反应中表现出较高活性,顺-1,4-聚合选择性高达97%,得到的聚异戊二烯具有荧光性。
同时,所述反应体系中,所述稀土金属配合物的支撑配体还会与烷基铝试剂的烷基发生置换反应,得到置换后的稀土金属烷基有机配合物和铝配合物,实现探测烯烃配位聚合反应的催化活性物种的荧光探针作用。
为实现上述目的,本发明采用以下技术方案。
一种双吡咯甲烯型稀土金属配合物,所述稀土金属配合物的结构如结构式Ⅰ和结构式Ⅱ所示:
其中,R
R
R
Ln是稀土金属,为钪(Sc)、钇(Y)、镧(La)、铈(Ce)、镨(Pr)、钕(Nd)、钷(Pm)、钐(Sm)、铕(Eu)、钆(Gd)、铽(Tb)、镝(Dy)、钬(Ho)、铒(Er)、铥(Tm)、镱(Yb)或镥(Lu)。
一种本发明所述双吡咯甲烯型稀土金属配合物的制备方法,所述方法步骤如下:
(1)制备双吡咯甲烯型配体
在保护气体氛围下将芳香醛和取代的吡咯加入到反应器中,加入二氯甲烷作溶剂,滴加加入三氟乙酸,反应2h~4h后,加入2,3-二氯-5,6-二氰基-1,4-苯醌(DDQ)进行氧化脱氢反应15min~30min,得到产物,将产物萃取、干燥以及过滤,将滤液用柱层析纯化,得到双吡咯甲烯型配体;
保护气体为氮气或惰性气体;
取代的吡咯中的取代基为所述R
芳香醛、取代的吡咯和DDQ的摩尔比为1:2:1.05~1.2,三氟乙酸摩尔量为芳香醛摩尔量的0.1%~0.5%;
优选将DDQ用二氯甲烷溶解后滴加加入;
优选柱层析使用的洗脱剂为二氯甲烷和甲醇的混合溶液,优选二氯甲烷与甲醇的体积比为99:1。
(2)制备双吡咯甲烯型稀土金属配合物
将步骤(1)制得的双吡咯甲烯型配体加入到反应器中,用甲苯作溶剂,得到混合物h;将混合物h滴加到溶解有金属源的甲苯溶液中,在20℃~25℃搅拌反应2h~6h,得到反应液;除去反应液中的溶剂得到固体,用正己烷洗涤3次~5次后,除去溶剂,得到的固体粉末用正己烷和四氢呋喃的混合溶剂或正己烷和甲苯的混合溶剂溶解,在-20℃~-35℃放置结晶,得到的晶体为本发明所述的双吡咯甲烯型稀土金属配合物;
步骤(2)在无水无氧环境下进行,具体可采用在手套箱中实现;
金属源为含有Ln的化合物,优选为二四氢呋喃-三(三甲基硅亚甲基)稀土金属化合物(Ln(CH2SiMe3)3(thf)2);
优选双吡咯甲烯型配体与金属源的摩尔比为1:1;
优选将混合物h置于手套箱中,冷冻后再滴加到溶解有金属源的甲苯溶液中,以减少反应放热对反应温度的影响。
一种本发明所述双吡咯甲烯型稀土金属配合物的应用,所述应用为将所述稀土金属配合物作为催化剂和荧光探针,与烷基铝试剂和有机硼盐一起用于:
(1)α-烯烃、环烯烃、共轭二烯烃、非共轭二烯烃、炔烃、极性单体或含杂原子的芳香烯烃的均聚反应;或
(2)α-烯烃、环烯烃、共轭二烯烃、非共轭二烯烃,炔烃、极性单体和含杂原子的芳香烯烃中任意两种的共聚反应;或
(3)α-烯烃、环烯烃、共轭二烯烃、非共轭二烯烃、炔烃、极性单体和含杂原子的芳香烯烃中的任一种与二氧化碳(CO2)的共聚反应;
有机硼盐与所述稀土金属配合物的摩尔比为1~2:1;
烷基铝试剂与所述稀土金属配合物的摩尔比为0.5~50:1;
α-烯烃、环烯烃、共轭二烯烃、非共轭二烯烃、炔烃、极性单体和含杂原子的芳香烯烃中任一种与所述稀土金属配合物的摩尔比为200~5000:1。
所述均聚反应步骤如下:
(1)向反应器中依次加入良溶剂、烷基铝试剂、所述稀土金属配合物、单体a和有机硼盐,-30℃~120℃搅拌反应0.1h~72h,得到置换后的稀土金属烷基配合物和铝配合物,以及聚合后的反应液;
步骤(1)在无水无氧条件下进行,可采用手套箱实现。
(2)向反应器中加入链终止剂,使反应中止,得到终止聚合反应的聚合物反应液;将聚合物反应液用无水乙醇进行沉降,析出固体物质,将固体物质在40℃~60℃真空干燥,除去溶剂至恒重,得到均聚产物;
烷基铝试剂、单体a、有机硼盐与所述稀土金属配合物的摩尔比为0.5~50:200~5000:1~2:1;
单体a为α-烯烃、环烯烃、共轭二烯烃、非共轭二烯烃、炔烃、极性单体或含杂原子的芳香烯烃。
所述均聚产物分布有置换后的铝配合物,因此具有荧光性。
所述共聚反应步骤如下:
(1)向反应器中依次加入良溶剂、烷基铝试剂、所述稀土金属配合物、单体b和有机硼盐,-60℃~120℃搅拌反应0.1h~72h,得到置换后的稀土金属烷基配合物和铝配合物,以及聚合后的反应液;
步骤(1)在无水无氧条件下进行,可采用手套箱实现。
(2)向反应器中加入链终止剂,使反应中止,得到终止聚合反应的聚合物反应液;将聚合物反应液用无水乙醇进行沉降,析出固体物质,将固体物质在40℃~60℃真空干燥,除去溶剂至恒重,得到共聚产物;
烷基铝试剂、单体b、有机硼盐和所述稀土金属配合物的摩尔比为0.5~50:200~5000:1~2:1;
单体b为α-烯烃、环烯烃、共轭二烯烃、非共轭二烯烃、炔烃、极性单体和含杂原子的芳香烯烃中的任两种;或
单体b为α-烯烃、环烯烃、共轭二烯烃、非共轭二烯烃、炔烃、极性单体和含杂原子的芳香烯烃中的任一种与二氧化碳。
所述均聚产物分布有置换后的铝配合物,因此具有荧光性。
所述均聚反应和共聚反应中:
优选良溶剂为正己烷、正庚烷、苯、甲苯、环己烷、氯苯、邻二氯苯、间二氯苯、对二氯苯、三氯苯和四氢呋喃中的一种以上。
烷基铝试剂是分子式为AlX3的烷基铝、分子式为HAlX2的烷基氢化铝、分子式为AlX2Cl的烷基氯化铝或铝氧烷,X为烷基。
优选烷基铝为三甲基铝、三乙基铝、三正丙基铝、三正丁基铝、三异丙基铝、三异丁基铝、三己基铝、三环己基铝或三辛基铝。
优选烷基氢化铝为氢化二甲基铝、氢化二乙基铝、氢化二正丙基铝、氢化二正丁基铝、氢化二异丙基铝、氢化二异丁基铝、氢化二戊基铝、氢化二己基铝、氢化二环己基铝或氢化二辛基铝。
优选烷基氯化铝为氯化二甲基铝、氯化二乙基铝、氯化二正丙基铝、氯化二正丁基铝、氯化二异丙基铝、氯化二异丁基铝、氯化二戊基铝、氯化二己基铝、氯化二环己基铝或氯化二辛基铝。
优选铝氧烷为甲基铝氧烷、乙基铝氧烷、正丙基铝氧烷或正丁基铝氧烷。
有机硼盐为三苯基(甲基)-四(五氟苯)硼盐([Ph3C][B(C6F5)4])、苯基-二甲基氨基-四(五氟苯)硼盐([PhMe2NH][B(C6F5)4])、苯基-二甲基氨基-四苯基硼盐([PhMe2NH][BPh4])或三(五氟苯)硼盐(B(C6F5)3)。
优选α-烯烃为乙烯、丙烯、1-丁烯、1-戊烯、4-甲基-1-戊烯、1-己烯、1-庚烯、1-辛烯、1-癸烯、1-十二碳烯、1-十四碳烯、1-十六碳烯、1-二十碳烯、苯乙烯、α-甲基苯乙烯或3-氯甲基苯乙烯。
优选环烯烃为降冰片烯、极性降冰片烯、降冰片二烯、亚乙基降冰片烯、苯基降冰片烯、乙烯基降冰片烯或双环戊二烯。
优选共轭二烯烃为1,3-丁二烯、异戊二烯、月桂烯、1,3-环己二烯、罗勒烯或环戊二烯。
优选非共轭二烯烃为间戊二烯、1,5-己二烯、1,6-庚二烯或1,7-辛二烯。
优选炔烃为乙炔、对苯乙二炔、二乙炔基芳烃或苯基乙炔。
优选极性单体为环氧烷烃、内酯或2-乙烯基吡啶。
优选环氧烷烃为环氧乙烷、环氧丙烷、1,2-环氧丁烷、2,3-环氧丁烷、异环氧丁烷、环氧氯丙烷、环氧溴丙烷或三氟环氧丙烷;
内酯为ε-己内酯、β-丁内酯、δ-戊内酯、丙交酯、乙交酯或3-甲基-乙交酯。
链终止剂为含有2,6-二叔丁基-4-甲基苯酚、2,3,4-三甲基苯酚、间二苯酚、2,6-二乙基苯酚或对叔丁基苯酚的乙醇溶液;优选2,6-二叔丁基-4-甲基苯酚、2,3,4-三甲基苯酚、间二苯酚、2,6-二乙基苯酚或对叔丁基苯酚的质量分数为5%~15%。
一种本发明所述双吡咯甲烯型稀土金属配合物在合成聚异戊二烯中的应用,当单体a为异戊二烯时,所述稀土金属配合物在催化异戊二烯聚合反应中表现出较高活性,顺-1,4-聚合选择性高达97%,得到的聚异戊二烯中分布有置换后的铝配合物,因此具有荧光性。
有益效果
1.本发明提供了一种双吡咯甲烯型稀土金属配合物,所述稀土金属配合物具有较高的催化活性和选择性;
2.本发明提供了一种双吡咯甲烯型稀土金属配合物的制备方法,所述方法以吡咯衍生物、芳香醛等为初始原料,原料价廉易得且易于修饰,经济效率高,环保性好,适合工业化生产;
3.本发明提供了一种双吡咯甲烯型稀土金属配合物的应用,所述稀土金属配合物作为催化剂,与烷基铝试剂和有机硼盐组成催化体系,可催化单体a的均聚反应或单体b的共聚反应得到一系列具有特定结构的新型聚合材料;
4.本发明提供了一种双吡咯甲烯型稀土金属配合物的应用,当单体a为异戊二烯时,所述稀土金属配合物在催化异戊二烯聚合反应中表现出高活性,顺-1,4-聚合选择性高达97%,获得了具有荧光性质的聚异戊二烯,有极大的应用价值;
5.本发明提供了一种双吡咯甲烯型稀土金属配合物的应用,所述稀土金属配合物具有红色荧光性(λem=590nm),可以作为置换型荧光探针探测烯烃配位聚合活性物种,具体为通过聚合过程中所述稀土金属配合物的双吡咯甲烯基配体与烷基铝的烷基发生置换反应,双吡咯甲烯基配体与铝结合,生成有绿色荧光(λem=510nm)的金属铝配合物,分布在得到的聚合物中,通过根据烯烃配位聚合反应过程中催化体系荧光信号的变化情况和原位核磁,可直观得到催化活性物种的基本结构信息,为证明该聚合体系的配位聚合机理提供了强有力的佐证。
具体反应原理如下:
在聚合反应过程中,通过紫外灯或荧光光谱仪可以观察到聚合反应液的颜色从红色转变成绿色,这表明在反应过程中所述稀土金属配合物的双吡咯甲烯基配体从稀土金属离子上转移到铝离子上面,最后得到置换后的金属铝配合物(绿色荧光)和置换后的稀土烷基催化活性物种;所述活性物种在有机硼盐助催化剂的活化下拨除一个烷基,还会残留两个金属-碳键等待烯烃单体的配位和插入,进而实现配位聚合反应,这是首次利用荧光探针清晰观察到主催化剂上的支撑配体转移到烷基铝助催化剂上面去,清晰明确地阐明了金属单烷基催化剂前体在烯烃配位聚合反应的催化活性物种的真实结构,为其他金属单烷基催化剂的设计提供理论上的依据。
附图说明
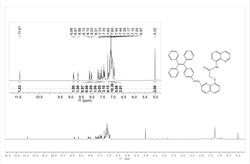
图1为实施例1中双吡咯甲烯配体(L1H)的核磁氢谱。
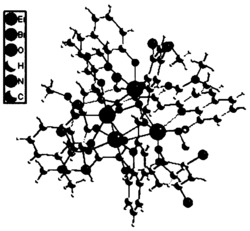
图2为实施例6中双吡咯甲烯型钪配合物(Sc-1)的X-ray表征晶体结构。
图3为实施例6中双吡咯甲烯型钪配合物(Sc-1)的核磁氢谱。
图4为实施例6中双吡咯甲烯型钪配合物(Sc-1)的核磁碳谱。
图5为实施例6中双吡咯甲烯型钪配合物(Sc-1)的荧光光谱。
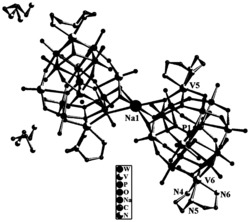
图6为实施例7中双吡咯甲烯型钇配合物(Y-1)的X-ray表征晶体结构。
图7为实施例8中双吡咯甲烯型镥配合物(Lu-1)的X-ray表征晶体结构。
图8为实施例9中终产物的核磁氢谱谱图。
图9为实施例9中终产物的核磁碳谱谱图。
图10为实施例9中终产物的GPC谱图。
图11为实施例9中终产物的荧光发射光谱图。
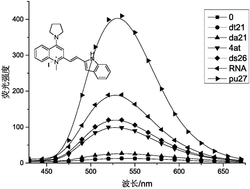
图12为实施例11中终产物的荧光发射光谱图。
图13为实施例16中终产物的核磁氢谱谱图。
图14为实施例16中终产物的核磁碳谱谱图。
图15为实施例16中终产物的GPC谱图。
图16为实施例17中终产物的核磁氢谱谱图。
图17为实施例17中终产物的GPC谱图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下将结合实施例进一步阐述本发明。
以下实施例中提到的主要试剂见表1,主要仪器和设备见表2。
表1主要试剂
表2主要仪器和设备
以下实施例中聚合活性由公式Activity=(m·yeild)/(ncat·time)计算得出。其中,Activity为聚合活性,单位为kg·mol
对实施例制备得到产物作为样品,采用核磁共振仪进行核磁共振检测:
产物的特征峰值及微观结构可以由
(1)1,4-聚异戊二烯的选择性(所占比例):
Mol 1,4-IP%={IH1/(IH1+0.5IH2)}×100
(2)3,4-聚异戊二烯的选择性(所占比例):
Mol 3,4-IP%={0.5IH2/(IH1+0.5IH2)}×100
(3)顺1,4-聚异戊二烯的选择性(所占比例):
Mol cis-1,4-IP%={IC1/(IC1+IC2+IC3)}×100
(4)反1,4-聚异戊二烯的选择性(所占比例):
Mol trans-1,4-IP%={IC3/(IC1+IC2+IC3)}×100
其中,IP为聚异戊二烯,IH1为
对实施例制备得到产物作为样品进行X-ray测试:
在显微镜下挑选晶体,晶体尺寸0.1mm~0.5mm,置于-100℃下低温测试;
对实施例制备得到产物作为样品进行凝胶渗透色谱(GPC)测试:
四氢呋喃作溶剂,流速为1ml/分钟,温度45℃,聚苯乙烯作参标;
对实施例制备得到所述稀土金属配合物、聚合物以及置换后的铝配合物作为样品进行荧光性质测试:甲苯或二氯甲烷作溶剂溶解样品,样品浓度为10
实施例1
在氮气氛围下将2,4,6-三甲基苯甲醛742mg(5mmol)和2,4-二甲基吡咯950mg(10mmol)溶于200ml重蒸的二氯甲烷中,滴加2滴三氟乙酸,三氟乙酸摩尔量为2,4,6-三甲基苯甲醛摩尔量的0.5%,室温反应2h,薄层层析(TLC)监测至2,4,6-三甲基苯甲醛反应完全;再向反应液中滴加含有1.2g DDQ(5.3mmo)的二氯甲烷溶液,反应30min后,向反应体系中加入120ml水,二氯甲烷萃取,合并有机相,用无水Na2SO4干燥后过滤,浓缩滤液,柱层析纯化,柱层析使用的洗脱剂为二氯甲烷和甲醇的混合溶液,二氯甲烷与甲醇的体积比为99:1,得到产物1.2g,产率为55%。
化学反应方程式如下:
对本实施例制得的产物进行核磁共振检测,结果如图1所示,具体为:
实施例2
在氮气氛围下将苯甲醛530mg(5mmol)和2,4-二甲基吡咯950mg(10mmol)溶于200ml重蒸的二氯甲烷中,滴加2滴三氟乙酸,三氟乙酸摩尔量为苯甲醛摩尔量的0.5%,室温反应2h,TLC监测至苯甲醛反应完全;再向反应液中滴加含有1.2g DDQ(5.3mmol)的二氯甲烷溶液,反应30min后,向反应体系中加入120ml水,二氯甲烷萃取,合并有机相,用无水Na2SO4干燥后过滤,浓缩滤液,柱层析纯化,柱层析使用的洗脱剂为二氯甲烷和甲醇的混合溶液,二氯甲烷与甲醇的体积比为99:1,得到产物500mg,产率为18%。
对本实施例制得的产物进行核磁共振检测,具体为:
实施例3
在氮气氛围下将对硝基苯甲醛755mg(5mmol)和2,4-二甲基吡咯950mg(10mmol)溶于200ml重蒸的二氯甲烷中,滴加2滴三氟乙酸,三氟乙酸摩尔量为对硝基苯甲醛摩尔量的0.5%,室温反应2h,TLC监测至对硝基苯甲醛反应完全;再向反应液中滴加含有1.2g DDQ(5.3mmol)的二氯甲烷溶液,反应30min后,向反应体系中加入120ml水,二氯甲烷萃取,合并有机相,用无水Na2SO4干燥后过滤,浓缩滤液,柱层析纯化,柱层析使用的洗脱剂为二氯甲烷和甲醇的混合溶液,二氯甲烷与甲醇的体积比为99:1,得到产物500mg,产率为31%。
对本实施例制得的产物进行核磁共振检测,具体为:
实施例4
在氮气氛围下将2,6-二甲基苯甲醛670mg(5mmol)和2,4-二甲基吡咯950mg(10mmol)溶于200ml重蒸的二氯甲烷中,滴加2滴三氟乙酸,三氟乙酸摩尔量为2,6-二甲基苯甲醛摩尔量的0.5%,室温反应2h,TLC监测至2,6-二甲基苯甲醛反应完全;再向反应液中滴加含有1.2g DDQ(5.3mmol)的二氯甲烷溶液,反应30min后,向反应体系中加入120ml水,二氯甲烷萃取,合并有机相,用无水Na2SO4干燥后过滤,浓缩滤液用柱层析纯化,柱层析使用的洗脱剂为二氯甲烷和甲醇的混合溶液,二氯甲烷与甲醇的体积比为99:1,得到产物650mg,产率为43%。
对本实施例制得的产物进行核磁共振检测,具体为:
实施例5
在氮气氛围下将对甲基苯甲醛600mg(5mmol)和2,4-二甲基吡咯950mg(10mmol)溶于200ml重蒸的二氯甲烷中,滴加2滴三氟乙酸,三氟乙酸摩尔量为对甲基苯甲醛摩尔量的0.5%,室温反应2h,TLC监测至对甲基苯甲醛反应完全;再向反应液中滴加含有1.2g DDQ(5.3mmol)的二氯甲烷溶液,反应30min后,向反应体系中加入120ml水,二氯甲烷萃取,合并有机相,用无水Na2SO4干燥后过滤,浓缩滤液用柱层析纯化,柱层析使用的洗脱剂为二氯甲烷和甲醇的混合溶液,二氯甲烷与甲醇的体积比为99:1,得到产物450mg,产率为31%。
对本实施例制得的产物进行核磁共振检测,具体为:
实施例6
在手套箱中,将实施例1制得的L1H 200mg(0.63mmol)加入到茄瓶中,用25ml甲苯作溶剂,充分溶解,得到混合物h,置于冰箱中冷冻0.5h;将Sc(CH2SiMe3)3(thf)2 283mg(0.63mmol)溶解在3mL甲苯中,将混合物h滴加到溶解有Sc(CH2SiMe3)3(thf)2的甲苯溶液中,在25℃搅拌反应4h;用真空泵将反应液抽干得到固体,用正己烷洗涤3次,每次5mL,用真空泵抽干除去溶剂,得到固体粉末用正己烷和四氢呋喃的混合溶剂溶解,混合溶剂中正己烷与四氢呋喃的体积比为10:3,在-30℃放置结晶后,得到晶体即产物,质量为120mg,产率为25%,整个反应都在手套箱的无水无氧环境下进行。
化学反应方程式如下:
对本实施例制得的产物进行X-ray表征,晶体结构如图2所示,晶体数据和结构:分子式:C48H61N4SiSc,分子量:767.05,晶系:单斜,空间群: α/°:90.00,β/°:106.57(3),γ/°:90.00, Z:4;具体为:键角:N1-Sc1-N3 93.76(6);N1-Sc1-N4170.27(5);N1-Sc1-C45,86.36(6);N2-Sc1-N1,83.61(5);N2-Sc1-N3,104.8(5);N2-Sc1-N4,105.45(5);N2-Sc1-C45,115.43(6);N3-Sc1-N4,80.74(6);N3-Sc1-C45,140.20(6);N4-Sc1-C45,92.76(6);键长:Sc1-N1,2.1883(14);Sc1-N2,2.1495(14);Sc1-N3,2.1919(14);Sc1-N4,2.1943(14);Sc1-C45,2.2605(18);Si1-C45,1.8396(19);Si1-C46,1.885(2);Si1-C47,1.876(2);Si1-C48,1.876(2)。
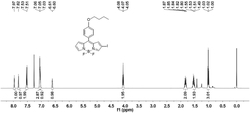
对本实施例制得的产物进行核磁共振检测,图3为所述产物的核磁氢谱,具体为:
图4为所述产物的核磁碳谱,具体为:
因此,本实施例制得的产物为一种双吡咯甲烯型稀土金属配合物,具体为双吡咯甲烯型钪配合物(简称为Sc-1)。
用荧光光谱仪对Sc-1进行荧光性质测试,图5为所述产物的荧光发射光谱,结果如下:λem=590nm,说明Sc-1可以发射红色荧光。
实施例7
制备双吡咯甲烯型稀土金属钇
在手套箱中,将实施例1制得的L1H 200mg(0.63mmol)加入到茄瓶中,用25ml甲苯作溶剂,充分溶解,得到混合物h,置于冰箱中冷冻0.5h;将Y(CH2SiMe3)3(thf)2 311mg(0.63mmol)溶解在3mL甲苯中,将混合物h滴加到溶有Y(CH2SiMe3)3(thf)2的甲苯溶液中,在25℃搅拌反应4h;用真空泵将反应液抽干得到固体,用正己烷洗涤3次,每次5mL,用真空泵抽干除去溶剂,得到固体粉末用正己烷和四氢呋喃的混合溶剂溶解,混合溶剂中正己烷与四氢呋喃的体积比为10:3,在-30℃放置结晶后,得到晶体即产物,质量为130mg,产率为20%,整个反应都在手套箱的无水无氧环境下进行。
化学反应方程式如下:
对本实施例制得的产物进行X-ray表征,晶体结构如图6所示。晶体数据和结构:分子式:C60H90N4OSi3Y2,分子量:1145.45,晶系:单斜,空间群: α/°:90.00,β/°:90.33(3),γ/°:90.00, Z:4;因此,本实施例制得的产物为一种双吡咯甲烯型稀土金属配合物,具体为双吡咯甲烯型钇配合物(简称为Y-1)。
用荧光光谱仪对Y-1进行荧光性质测试,所述产物的荧光发射光谱与图5类似,结果如下:λem=590nm,说明Y-1可以发射红色荧光。
实施例8
在手套箱中,将实施例1制得的L1H 200mg(0.63mmol)加入到茄瓶中,用25ml甲苯作溶剂,充分溶解,得到混合物h,置于冰箱中冷冻0.5h;将Lu(CH2SiMe3)3(thf)2 364mg(2mmol)溶解在3mL甲苯中,将混合物h滴加到溶有Lu(CH2SiMe3)3(thf)2的甲苯溶液中,在25℃搅拌反应2h;用真空泵将反应液抽干得到固体,用正己烷洗涤3次,每次5mL,用真空泵抽干除去溶剂,得到固体粉末,得到固体粉末用正己烷和四氢呋喃的混合溶剂溶解,混合溶剂中正己烷与四氢呋喃的体积比为10:3,在-30℃放置结晶后,得到晶体即产物,质量为150mg,产率为27%,整个反应都在手套箱的无水无氧环境下进行。
化学反应方程式如下:
对本实施例制得的产物进行X-ray表征,晶体结构如图7所示,可知晶体数据和结构:分子式:C60H90N4OLu2Si3,分子量:1317.56,晶系:三斜晶系,空间群:P-1, α/°:62.42(3),β/°:74.99(3),γ/°:64.88(3), Z:4;因此,本实施例制得的产物为一种双吡咯甲烯型稀土金属配合物,具体为双吡咯甲烯型镥配合物(简称为Lu-1)。
用荧光光谱仪对Lu-1进行荧光性质测试,测试结果与Y-1相同。
实施例9
(1)将茄瓶置于手套箱中,向茄瓶中依次加入5mL甲苯、200μmol Al
(2)向反应器中加入30mL含有质量分数为5%的2,6-二叔丁基-4-甲基苯酚的乙醇溶液,使反应中止,得到终止聚合反应的聚合物反应液;将聚合物反应液用乙醇进行沉降,析出红色固体物质,将所述固体物质在40℃下真空干燥,除去溶剂至恒重,得到终产物,净重0.27g。
对本实施例中终产物分别进行如下测试:
(1)核磁共振
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁氢谱如图8所示,具体为:δ=5.13,4.72ppm;
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁碳谱如图9所示,具体为:δ=23.4,26.4,32.2,125.0,135.2ppm;
由核磁谱图分析可以判断所述终产物为聚异戊二烯,产率为100%,顺-1,4-聚合选择性为95%,聚合活性为81×10
(2)凝胶渗透色谱(GPC)
本实施例制备的终产物的凝胶渗透色谱图谱见图10,由谱图结果Mn和Mw分析可知,终产物的数均分子量Mn=137×10
(3)用荧光光谱仪对本实施例制备的终产物,进行荧光性质测试,结果如下:
本实施例的荧光发射光谱见图11,由谱图结果可知,终产物聚异戊二烯的发射波长为508nm。
因为聚合反应过程中Sc-1中的双吡咯甲烯基配体与烷基铝的烷基发生置换反应,生成新的稀土金属烷基配合物,伴随生成了具有绿色荧光性的铝配合物,置换后生成的铝配合物均匀的掺杂于聚异戊二烯中,所有得到具有荧光性的聚异戊二烯,同时也证明所述稀土金属配合物可以作为置换型荧光探针探测烯烃配位聚合活性物种。
实施例10
(1)将茄瓶置于手套箱中,向茄瓶中依次加入5mL甲苯、200μmol Al
(2)加入30mL含有质量分数为5%的2,6-二叔丁基-4-甲基苯酚的乙醇溶液,使反应中止,得到终止聚合反应的聚合物反应液;将聚合物反应液用乙醇进行沉降,析出红色固体物质,将固体物质在40℃下真空干燥,除去溶剂至恒重,得到终产物,净重0.27g。
对本实施例中终产物分别进行如下测试:
(1)核磁共振
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁氢谱与图8类似:δ=5.13,4.72ppm;
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁碳谱与图9类似:δ=23.4,26.4,32.2,125.0,135.2ppm;
由核磁谱图可以看出所述终产物为聚异戊二烯,产率为100%,顺-1,4-聚合选择性为85%,聚合活性为9×10
(2)凝胶渗透色谱
本实施例制备的终产物的凝胶渗透色谱图谱与图10类似,由谱图结果Mn和Mw分析可知,终产物的数均分子量Mn=76×10
(3)用荧光光谱仪对本实施例制备的终产物,进行荧光性质测试,结果与实施例9结果相同。
实施例11
(1)将茄瓶置于手套箱中,向茄瓶中依次加入5mL甲苯、200μmol AlMe3、20μmolSc-1、4mmol异戊二烯及20μmol[Ph3C][B(C6F5)4],在25℃搅拌反应10min,得到置换后的荧光探针和正在进行聚合的反应液。
(2)向反应器中加入30mL含有质量分数为5%的2,6-二叔丁基-4-甲基苯酚的乙醇溶液,使反应中止,得到终止聚合反应的聚合物反应液;将聚合物反应液用乙醇进行沉降,析出红色固体物质,将所述固体物质在40℃下真空干燥,除去溶剂至恒重,得到终产物,净重0.25g。
对本实施例中终产物分别进行如下测试:
(1)核磁共振
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁氢谱与图8类似:δ=5.13,4.72ppm;
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁碳谱与图9类似:δ=23.4,26.4,32.2,125.0,135.2ppm;
由核磁谱图可以看出所述终产物为聚异戊二烯,产率为92%,顺-1,4-聚合选择性为97%,聚合活性为0.5×10
(2)凝胶渗透色谱
本实施例制备的终产物的凝胶渗透色谱图谱与图10类似,由谱图结果Mn和M分析可知,终产物的数均分子量Mn=17×10
(3)用荧光光谱仪对本实施例制备的终产物进行荧光性质测试,结果如图12所示,发射波长为510nm。
实施例12
(1)将茄瓶置于手套箱中,向茄瓶中依次加入5mL甲苯、200μmol Al
(2)加入30mL含有质量分数为5%的2,6-二叔丁基-4-甲基苯酚的乙醇溶液,使反应中止,得到终止聚合反应的聚合物反应液;将聚合物反应液用乙醇进行沉降,析出红色固体物质,将所述固体物质在40℃下真空干燥,除去溶剂至恒重,得到终产物,净重0.27g。
对本实施例中终产物分别进行如下测试:
(1)核磁共振
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁氢谱与图8类似,具体为:δ=5.13,4.72ppm;
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁碳谱与图9类似,具体为:δ=23.4,26.4,32.2,125.0,135.2ppm;
由核磁谱图可以看出所述终产物为聚异戊二烯,产率为100%,顺-1,4聚合选择性为90%,聚合活性为0.8×10
(2)凝胶渗透色
本实施例制备的终产物的凝胶渗透色谱图谱与图10类似,由谱图结果Mn和Mw分析可知,聚异戊二烯的数均分子量Mn=62×10
(3)用荧光光谱仪对本实施例制备的终产物,进行荧光性质测试,结果与实施例9结果相同。
实施例13
(1)将茄瓶置于手套箱中,向茄瓶中依次加入5mL甲苯、200μmol Al
(2)加入30mL含有质量分数为5%的2,6-二叔丁基-4-甲基苯酚的乙醇溶液,使反应中止,得到终止聚合反应的聚合物反应液;将聚合物反应液用乙醇进行沉降,析出红色固体物质,将固体物质在40℃下真空干燥,除去溶剂至恒重,得到终产物,净重0.27g。
对本实施例中终产物分别进行如下测试:
(1)核磁共振
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁氢谱与图8类似,具体为:δ=5.13,4.72ppm;
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁碳谱与图9类似,具体为:δ=23.4,26.4,32.2,125.0,135.2ppm;
由核磁谱图可以看出所述终产物为聚异戊二烯,产率为100%,顺-1,4-聚合选择性为87%,聚合活性为0.8×10
(2)凝胶渗透色谱
本实施例制备的终产物的凝胶渗透色谱图谱与图10类似,由谱图结果Mn和Mw分析可知,终产物的数均分子量Mn=65×10
(3)用荧光光谱仪对本实施例制备的终产物,进行荧光性质测试,结果与实施例9结果相同。
实施例14
(1)将茄瓶置于手套箱中,向茄瓶中依次加入5mL甲苯、200μmol Al
(2)加入30mL含有质量分数为5%的2,6-二叔丁基-4-甲基苯酚的乙醇溶液,使反应中止,得到终止聚合反应的聚合物反应液;将聚合物反应液用乙醇进行沉降,析出固体物质,将固体物质在40℃下真空干燥,除去溶剂至恒重,得到终产物,净重1.35g。
对本实施例中终产物分别进行如下测试:
(1)核磁共振
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁氢谱与图8类似,具体为:δ=5.13,4.72ppm;
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁碳谱与图9类似,具体为:δ=23.4,26.4,32.2,125.0,135.2ppm;
由核磁谱图可以看出所述终产物为聚异戊二烯,产率为100%,顺-1,4-聚合选择性为91%,聚合活性为405×10
(2)凝胶渗透色谱
本实施例制备的终产物的凝胶渗透色谱图谱与图10类似,由谱图结果Mn和Mw分析可知,终产物的数均分子量Mn=185×10
(3)用荧光光谱仪对本实施例制备的终产物,进行荧光性质测试,结果与实施例9结果相同。
实施例15
(1)将茄瓶置于手套箱中,向茄瓶中依次加入5mL甲苯、200μmol Al
(2)加入30mL含有质量分数为5%的2,6-二叔丁基-4-甲基苯酚的乙醇溶液,使反应中止,得到终止聚合反应的聚合物反应液;将聚合物反应液用乙醇进行沉降,析出固体物质,将固体物质在40℃下真空干燥,除去溶剂至恒重,得到终产物,净重2.7g。
对本实施例中终产物分别进行如下测试:
(1)核磁共振
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁氢谱与图8类似,具体为:δ=5.13,4.72ppm;
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁碳谱与图9类似,具体为:δ=23.4,26.4,32.2,125.0,135.2ppm;
由核磁谱图可以看出所述终产物为聚异戊二烯,产率为100%,顺-1,4-聚合选择性为86%,聚合活性为810×10
(2)凝胶渗透色谱
本实施例制备的终产物的凝胶渗透色谱图谱与图10类似,由谱图结果Mn和Mw分析可知,终产物的数均分子量Mn=276×10
(3)用荧光光谱仪对本实施例制备的终产物,进行荧光性质测试,结果与实施例9结果相同。
实施例16
(1)将茄瓶置于手套箱中,向茄瓶中依次加入10mL甲苯、200μmol Al
(2)加入30mL含有质量分数为5%的2,6-二叔丁基-4-甲基苯酚的乙醇溶液,使反应中止,得到终止聚合反应的聚合物反应液;将聚合物反应液用乙醇进行沉降,析出固体物质,将固体物质在40℃下真空干燥,除去溶剂至恒重,得到终产物,净重0.81g。
对本实施例中终产物分别进行如下测试:
(1)核磁共振
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁氢谱如图13所示,具体为:δ=5.13,5.11,4.72,ppm;
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁碳谱如图14所示,具体为:δ=23.4,26.4,29.7,32.2,37.09,37.51,42.18,125.0,135.2ppm;
由核磁谱图可以看出所述终产物为聚异戊二烯和月桂烯共聚物,产率为100%,顺-1,4-聚合选择性为86%,聚合活性为14×10
(2)凝胶渗透色谱
本实施例制备的终产物的凝胶渗透色谱图谱见图15,由谱图结果Mn和Mw分析可知,终产物的数均分子量Mn=41×10
(3)用荧光光谱仪对本实施例制备的终产物,进行荧光性质测试,结果仍与实施例9结果相同。
实施例17
(1)将茄瓶置于手套箱中,向茄瓶中依次加入10mL甲苯、200μmol Al
(2)加入30mL含有质量分数为5%的2,6-二叔丁基-4-甲基苯酚的乙醇溶液,使反应中止,得到终止聚合反应的聚合物反应液;将聚合物反应液用乙醇进行沉降,析出固体物质,将固体物质在40℃下真空干燥,除去溶剂至恒重,得到终产物,净重0.3g。
对本实施例中置换后荧光探针和终产物分别进行如下测试:
(1)核磁共振
对本实施例制得的终产物进行核磁共振检测,所述终产物的核磁氢谱如图16所示,具体为:δ=5.13-5.10,1.25-0.5,ppm;
由核磁谱图可以看出所述终产物为聚1,5-己二烯,产率为95%,成环率为100%,反-1,4选择性为70%,聚合活性为4.7×10
(2)凝胶渗透色谱
本实施例制备的终产物的凝胶渗透色谱图谱见图17,由谱图结果Mn和Mw分析可知,终产物的数均分子量Mn=2×10
(3)用荧光光谱仪对本实施例制备的终产物,进行荧光性质测试,结果与实施例9结果相同。
本发明包括但不限于以上实施例,凡是在本发明精神的原则之下进行的任何等同替换或局部改进,都将视为在本发明的保护范围之内。
一种双吡咯甲烯型稀土金属配合物、制备方法及应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。














动态评分
0.0