








专利摘要
本发明公开了一种二聚萘醌类衍生物的合成方法:式I所示的化合物、碱性物质在乙腈溶剂中,空气或者氧气气氛下,50‑100℃温度下搅拌反应10‑24小时,反应结束后,反应液后处理制备得到式II所示的二聚萘醌类衍生物。本发明的合成方法具有原料廉价易得、无过渡金属催化,氧源为空气,易得且环境友好,反应条件温和,高效合成目标底物且官能团普适性好、操作简便等优点。
说明书
(一)技术领域
本发明属于有机化合物合成技术领域,具体涉及一种二聚萘醌类衍生物的合成方法。
(二)背景技术
萘醌类化合物是一类广泛存在于自然界中的小分子化合物,由于其具有多样生物活性,被广泛应用于医药、农药和染料等多种领域。二聚萘醌是萘醌类化合物的一种衍生物,是由两分子萘醌类化合物通过C-C键连接形成。二聚萘醌类化合物由于其特殊的结构,具有着广泛的药理作用。生理学活性研究表明,二聚萘醌类化合物具有泻下、抗肿瘤、抗突变与致突变、抗炎、抗病毒与抗真菌、抗菌、抗原虫、抗氧化等活性,其代表物有3-3’-双胡桃醌、柿醌、异柿醌,其中后二者具有对肿瘤细胞对毒性和抗菌活性,有望成为新的抗癌药物。总之,关于萘醌类化合物的合成有很大的研究价值。
二聚萘醌类化合物是天然产物中的一员,其通常来源于对动植物体内成分的分离提纯,关于其人工合成鲜有报道。而以二聚萘醌或萘醌为原料,通过C-O键切断氧化合成二聚萘醌类化合物是应用较为广泛的路线,但其局限性太大,且工艺复杂,需要一系列昂贵的氧化剂、贵金属,且环境污染严重。(参见Takeya等人Tetrahedron,2004,60(47),10681-10693;Laatsch等人Liebigs Ann.Chem.,1990,(5),433-40等)。
鉴于以上存在的问题,发展一种廉价原料、无金属催化的、低廉的氧化剂氧化的、反应条件温和的且收率较高的合成二聚萘醌及其衍生物的方法迫在眉睫。
(三)发明内容
发明目的:针对现有技术中存在的不足,本发明旨在提供一种制备二聚萘醌及其衍生物的方法,克服现有技术的缺点,以廉价易得的、非醌类的1-(2-乙酰苯基)-2-苯乙二酮类化合物为原料、无需金属催化、以方便易得且环境友好的空气为氧源、实现温和高效的反应。
为了达到上述发明目的,本发明采用的技术方案是:
一种式II所示的二聚萘醌类衍生物的合成方法,所述方法为,式I所示的化合物、碱性物质在乙腈溶剂中,空气或者氧气气氛下,50-100℃温度下搅拌反应10-24小时,反应结束后,反应液后处理制备得到式II所示的二聚萘醌类衍生物;
式I或式II中,两个苯环上的H不被取代或各自独立的被单取代基R1、R2取代,所述R1、R2各自独立为甲基、乙基、丙基、甲氧基、乙氧基、氟、氯、溴或硝基,优选为甲基、丙基、氟或氯。
进一步,优选R1为氟或氯,更优选R1为3-F、4-F或4-Cl。
优选R2为甲基、丙基或氯,更优选R2为2-甲基、3-甲基、4-甲基、3-Cl、4-Cl或4-丙基。
式II中,不同位置的R1表示相同的取代情况;不同位置的R2表示相同的取代情况。
所述碱性物质为乙酸钾、甲酸钾、碳酸钾或碳酸钠,优选为乙酸钾。
所述碱性物质的物质的量用量为式I所示的化合物的物质的量的0.5-2倍,优选1倍。
所述反应温度为50-100℃,优选为60-80℃,更优选为70~80℃。
所述乙腈的体积用量通常以式I所示的化合物的物质的量计为5~50mL/mmol,优选为10~20mL/mmol。
本发明所述反应的时间为10~24小时,优选12~14小时,最优选12小时。
本发明反应在空气或者氧气气氛下进行,是以空气或氧气为氧源来进行反应,优选在空气中反应,较为节约成本。
本发明所述反应液后处理方法为:反应结束后,所得反应液中加入柱层析硅胶,减压蒸馏除去溶剂,剩余混合物装柱,经柱层析分离,以石油醚、乙酸乙酯体积比为6:1的混合溶剂作为洗脱剂,收集含有产物的洗脱液,洗脱液蒸除溶剂得到式II所示的二聚萘醌类衍生物。
本发明使用的原料1-(2-乙酰苯基)-2-苯乙二酮类化合物,本领域技术人员可以根据现有文献公开的方法自行制备,例如文献[Dmitry A.Gorlushko.Tetrahedron Lett.,2008,49(6),1080-1082;Manojveer.Seetharaman.Balamurugan,Org.Lett.,2014,16(6),1712-1715;Zhang,Wenxia.Synlett,2013,24(20),2709-2714.]等。
本发明原料廉价易得、无需金属催化剂、以方便易得且环境友好的空气为氧源,高收率合成二聚萘醌类产物。
较为具体的,推荐本发明所述方法按以下步骤进行:式I所示的化合物为原料,在乙酸钾作用下,在乙腈溶剂中,空气气氛下,50-100℃下搅拌反应10~24 小时,反应结束后,反应液中加入柱层析硅胶,减压蒸馏除去溶剂,剩余混合物装柱,经柱层析分离,以石油醚、乙酸乙酯体积比为6:1的混合溶剂作为洗脱剂,收集含有产物的洗脱液,洗脱液蒸除溶剂得到式II所示的二聚萘醌类衍生物,所述乙酸钾的物质的量用量为式I所示的化合物的物质的量的1倍。
本发明通过1-(2-乙酰苯基)-2-苯乙二酮类化合物在碱性物质作用下,以空气为氧源,通过亲核关环氧化偶联制得二聚萘醌及其衍生物,有益效果在于:与现有二聚萘醌及其衍生物的制备方法相比,原料廉价易得,无需催化剂,使用空气为氧化剂,成本大大降低,环境友好;反应条件温和,节约能源消耗;此外,还具有产率高,底物普适性强,操作简便等特点。
(四)具体实施方式
下面结合具体实施例对本发明作进一步详细说明,但本发明的保护范围不限于此:
实施例1
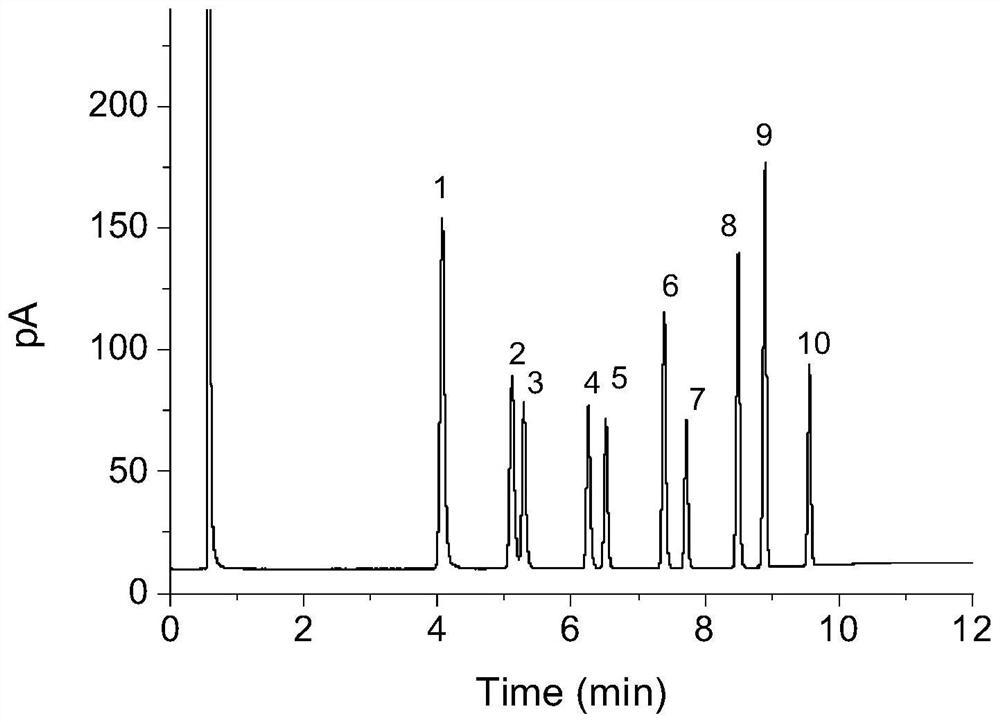
将50.4mg(0.2mmol)1-(2-乙酰苯基)-2-苯乙二酮、19.6mg(0.2mmol)乙酸钾、加入到10mL圆底烧瓶中,再加入2mL乙腈作溶剂。接着,于70℃下磁力搅拌12h。然后,在反应液中加入1g柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,剩余物装柱,通过柱色谱分离,以石油醚/乙酸乙酯 (V:V=6:1)作为洗脱剂,收集含有产物的洗脱液,洗脱液蒸除溶剂得到产物纯品3,3’-二苯基-2,2’-联萘-1,1’,4,4’-四酮。该物质为黄色固体,产率81%。表征数据:1H NMR(CDCl3,500MHz):δ8.23-8.21(m,1H),8.13-8.11(m,1H),7.85-7.79(m,2H),7.32-7.29(m,1H),7.20(t,J=8Hz,2H),6.69(d,J=7Hz,2H);13C NMR(CDCl3,125MHz):δ184.7,183.5,146.4,142.1,134.2,133.9,132.1,132.0,131.9,129.3,129.2,127.7,127.0,126.7.
实施例2
将57.3mg(0.2mmol)1-(2-乙酰苯基)-2-(3-氯苯基)乙二酮、16.8mg(0.2mmol)甲酸钾、加入到10mL圆底烧瓶中,再加入2mL乙腈作溶剂。接着,于80℃下磁力搅拌14h。然后,在反应液中加入1g柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,剩余物装柱,通过柱色谱分离,以石油醚/乙酸乙酯(V:V=6:1)作为洗脱剂,收集含有产物的洗脱液,洗脱液蒸除溶剂得到产物纯品3,3’-二(3-氯苯基)-2,2’-联萘-1,1’,4,4’-四酮。该物质为黄色固体,产率85%。
表征数据:1H NMR(CDCl3,500MHz):δ8.23-8.21(m,1H),8.15-8.13(m,1H),7.85-7.83(m,2H),7.34-7.31(m,1H),7.19(t,J=8Hz,1H),6.68-6.65(m,2H);13C NMR(CDCl3,125MHz):δ184.4,182.9,145.1,14 2.2,134.5,134.2,134.0,133.6,131.9,131.7,129.5,129.4,129.2,127.1,127.0,126.9.
实施例3
将53.2mg(0.2mmol)1-(2-乙酰苯基)-2-(2-甲基苯基)乙二酮、13.8mg(0.1mmol)碳酸钾、加入到10mL圆底烧瓶中,再加入3mL乙腈作溶剂。接着,于50℃下磁力搅拌24h。然后,在反应液中加入1g柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,剩余物装柱,通过柱色谱分离,以石油醚/乙酸乙酯(V:V=6:1)作为洗脱剂,收集含有产物的洗脱液,洗脱液蒸除溶剂得到产物纯品3,3’-二(2-甲基苯基)-2,2’-联萘-1,1’,4,4’-四酮。该物质为黄色固体,产率82%。
表征数据:1H NMR(CDCl3,500MHz):δ8.24-8.22(m,1H),8.13-8.11(m,1H),7.83-7.80(m,2H),7.23-7.20(m,1H),7.11(d,J=7.5Hz,1H),7.03(t,J=7.5Hz,1H),6.94(dd,J1=8Hz,J2=1Hz,1H),1.19(s,3H);13C NM R(CDCl3,125MHz):δ184.9,182.5,147.5,142.3,137.7,134.2,133.9,132.2,131.8,130.5,129.4,128.0,127.1,126.8,125.7,19.8.
实施例4
将53.2mg(0.2mmol)1-(2-乙酰苯基)-2-(4-甲基苯基)乙二酮、39.2mg(0.4mmol)乙酸钾、加入到10mL圆底烧瓶中,再加入4mL乙腈作溶剂。接着,于100℃下磁力搅拌10h。然后,在反应液中加入1g柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,剩余物装柱,通过柱色谱分离,以石油醚/乙酸乙酯(V:V=6:1)作为洗脱剂,收集含有产物的洗脱液,洗脱液蒸除溶剂得到产物纯品3,3’-二(4-甲基苯基)-2,2’-联萘-1,1’,4,4’-四酮。该物质为黄色固体,产率73%。
表征数据:1H NMR(CDCl3,500MHz):δ8.22-8.20(m,1H),8.13-8.11(m,1H),7.83-7.78(m,2H),7.01(d,J=8Hz,2H),6.61(d,J=8Hz,2H),2.32(s,3H);13C NMR(CDCl3,125MHz):δ184.7,183.7,146.5,141.9,139.2,134.1,133.8,132.1,132.0,129.2(2C),128.4,126.9,126.7,21.3.
实施例5
将57.3mg(0.2mmol)1-(2-乙酰苯基)-2-(4-氯苯基)乙二酮、10.6mg(0.1mmol)碳酸钾、加入到10mL圆底烧瓶中,再加入3mL乙腈作溶剂。接着,于80℃下磁力搅拌14h。然后,在反应液中加入1g柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,剩余物装柱,通过柱色谱分离,以石油醚/乙酸乙酯(V:V=6:1)作为洗脱剂,收集含有产物的洗脱液,洗脱液蒸除溶剂得到产物纯品3,3’-二(4-氯苯基)-2,2’-联萘-1,1’,4,4’-四酮。该物质为黄色固体,产率90%。
表征数据:1H NMR(CDCl3,500MHz):δ8.22-8.20(m,1H),8.14-8.12(m,1H),7.84-7.82(m,2H),7.21(d,J=8.5Hz,2H),6.68(dd,J1=6.5Hz,J2=2Hz,2H),2.32(s,3H);13C NMR(CDCl3,125MHz):δ184.4,18 3.0,145.3,142.0,135.6,134.5,134.2,131.8,131.7,130.6,130.3,128.1,127.1,126.8.
实施例6
将53.2mg(0.2mmol)1-(2-乙酰苯基)-2-(3-甲基苯基)乙二酮、29.4mg(0.3mmol)乙酸钾、加入到10mL圆底烧瓶中,再加入3mL乙腈作溶剂。接着,于60℃下磁力搅拌12h。然后,在反应液中加入1g柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,剩余物装柱,通过柱色谱分离,以石油醚/乙酸乙酯(V:V=6:1)作为洗脱剂,收集含有产物的洗脱液,洗脱液蒸除溶剂得到产物纯品3,3’-二(3-甲基苯基)-2,2’-联萘-1,1’,4,4’-四酮。该物质为黄色固体,产率80%。
表征数据:1H NMR(CDCl3,500MHz):δ8.23-8.21(m,1H),8.12-8.11(m,1H),7.84-7.79(m,2H),7.12-7.07(m,2H),6.59(d,J=7Hz,1H),6.45(s,1H),2.22(s,3H);13C NMR(CDCl3,125MHz):δ184.7,183.6,146.4,142.0,137.1,134.1,133.9,132.0,131.8,130.0,129.9,127.5,126.9,126.6,126.0,21.4.
实施例7
将54.0mg(0.2mmol)1-(2-乙酰-4-氟苯基)-2-苯乙二酮、21.2mg(0.2mmol)碳酸钾、加入到10mL圆底烧瓶中,再加入2mL乙腈作溶剂。接着,于100℃下磁力搅拌22h。然后,在反应液中加入1g柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,剩余物装柱,通过柱色谱分离,以石油醚/乙酸乙酯(V:V=6:1)作为洗脱剂,收集含有产物的洗脱液,洗脱液蒸除溶剂得到产物纯品7,7’-二氟-3,3’-二苯基-2,2’-联萘-1,1’,4,4’-四酮。该物质为黄色固体,产率63%。
表征数据:1H NMR(CDCl3,500MHz):δ8.16(dd,J1=9Hz,J2=5Hz,1H),7.85(dd,J1=8.5Hz,J2=2.5Hz,1H),7.48-7.44(m,1H),7.33-7.30(m,1H),7.21(t,J=8Hz,2H),6.68(d,J=7.5Hz,2H);13C NMR(CDCl3,125MHz):δ183.5(d,J=1.3Hz),182.1,166.2(d,J=257.5Hz),146.6,141.7(d,J=1.3Hz),134.3(d,J=7.5Hz),131.6,130.4(d,J=8.8Hz),129.4,12 9.2,128.5(d,J=3.8Hz),127.7,121.5(d,J=22.5Hz),113.4(d,J=23.8Hz).
实施例8
将54.0mg(0.2mmol)1-(2-乙酰-3-氟苯基)-2-苯乙二酮、16.8mg(0.2mmol)甲酸钾、加入到10mL圆底烧瓶中,再加入2mL乙腈作溶剂。接着,于80℃下磁力搅拌12h。然后,在反应液中加入1g柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,剩余物装柱,通过柱色谱分离,以石油醚/乙酸乙酯(V:V=6:1)作为洗脱剂,收集含有产物的洗脱液,洗脱液蒸除溶剂得到产物纯品8,8’-二氟-3,3’-二苯基-2,2’-联萘-1,1’,4,4’-四酮。该物质为黄色固体,产率69%。
表征数据:1H NMR(CDCl3,500MHz):δ7.96(dd,J1=8Hz,J2=1Hz,1H),7.79-7.75(m,1H),7.53-7.49(m,1H),7.34-7.31(m,1H),7.21(t,J=8Hz,2H),6.71-6.70(m,2H);13C NMR(CDCl3,125MHz):δ182.5,182.4(d,J=3.4Hz),160.9(d,J=267.5Hz),145.5,142.9,135.7(d,J=10Hz),133.6,131.4,129.3,129.2,127.8,123.4(d,J=2.5Hz),122.9(d,J=21.3Hz),119.6(d,J=5Hz).
实施例9
将57.3mg(0.2mmol)1-(2-乙酰-4-氯苯基)-2-苯乙二酮、9.8mg(0.1mmol)乙酸钾、加入到10mL圆底烧瓶中,再加入2mL乙腈作溶剂。接着,于100℃下磁力搅拌10h。然后,在反应液中加入1g柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,剩余物装柱,通过柱色谱分离,以石油醚/乙酸乙酯(V:V=6:1)作为洗脱剂,收集含有产物的洗脱液,洗脱液蒸除溶剂得到产物纯品7,7’-二氯-3,3’-二苯基-2,2’-联萘-1,1’,4,4’-四酮。该物质为黄色固体,产率85%。
表征数据:1H NMR(CDCl3,500MHz):δ8.16(d,J=2Hz,1H),8.06(d,J=8.5Hz,1H),7.75(dd,J1=8.5Hz,J2=2.5Hz,1H),7.32(t,J=7.5Hz,1H),7.21(t,J=8Hz,2H),6.67(d,J=7Hz,2H);13C NMR(CDCl3,125MHz):δ183.5,182.4,146.6,141.5,141.1,134.3,132.8,131.6,130.1,129.4,12 9.1,128.7,127.7,126.6.
实施例10
将56.8mg(0.2mmol)1-(2-乙酰-4-氟苯基)-2-(4-甲基苯基)乙二酮、27.6mg(0.2mmol)碳酸钾、加入到10mL圆底烧瓶中,再加入4mL乙腈作溶剂。接着,于50℃下磁力搅拌24h。然后,在反应液中加入1g柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,剩余物装柱,通过柱色谱分 离,以石油醚/乙酸乙酯(V:V=6:1)作为洗脱剂,收集含有产物的洗脱液,洗脱液蒸除溶剂得到产物纯品7,7’-二氟-3,3’-二(4-甲基苯基)-2,2’-联萘-1,1’,4,4’-四酮。该物质为黄色固体,产率85%。
表征数据:1H NMR(CDCl3,500MHz):δ8.16(dd,J1=8.5Hz,J2=5Hz,1H),7.85(dd,J1=8.5Hz,J2=2.5Hz,1H),7.47-7.44(m,1H),7.02(d,J=8Hz,2H),6.59(d,J=8Hz,2H),2.33(s,3H);13C NMR(CDCl3,125MHz):δ183.5(d,J=0.6Hz),182.4,166.2(d,J=256.3Hz),146.8,141.6(d,J=1.6Hz),139.5,134.4(d,J=7.9Hz),130.3(d,J=8.9Hz),129.1,128.8,1 28.6(d,J=3.1Hz),128.5,121.4(d,J=22.4Hz),113.3(d,J=23.4Hz),21.4.
实施例11
将61.6mg(0.2mmol)1-(2-乙酰-4-氟苯基)-2-(4-丙基苯基)乙二酮、19.6mg(0.2mmol)乙酸钾、加入到10mL圆底烧瓶中,再加入3mL乙腈作溶剂。接着,于70℃下磁力搅拌19h。然后,在反应液中加入1g柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,剩余物装柱,通过柱色谱分离,以石油醚/乙酸乙酯(V:V=6:1)作为洗脱剂,收集含有产物的洗脱液,洗脱液蒸 除溶剂得到产物纯品7,7’-二氟-3,3’-二(4-丙基苯基)-2,2’-联萘-1,1’,4,4’-四酮。该物质为黄色固体,产率82%。
表征数据:1H NMR(CDCl3,500MHz):δ8.17(dd,J1=8.5Hz,J2=5Hz,1H),7.84(dd,J1=8Hz,J2=2.5Hz,1H),7.48-7.44(m,1H),6.99(d,J=8Hz,2H),6.57(d,J=8Hz,2H),2.55(t,J=7.5Hz,2H),1.66-1.59(m,2H),0.94(t,J=7.5Hz,3H);13C NMR(CDCl3,125MHz):δ183.6(d,J=1.1Hz),182.4,166.2(d,J=256.3Hz),146.6,144.2,141.6(d,J=1.6Hz),134.4(d,J=8.8Hz),130.3(d,J=8.8Hz),129.2,129.0,128.6(d,J=3.1Hz),127.8,121.4(d,J=22.4Hz),113.3(d,J=23.4Hz),37.8,24.3,13.8。
一种二聚萘醌类衍生物的合成方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。


















动态评分
0.0