










专利摘要
本发明公开了一种氨糖噻唑衍生物及其合成方法。合成时以氨基葡萄糖盐酸盐为原料,对羟基进行苄基醚保护,实现了氨基的选择性反应,合成了新型中间体糖基硫脲—N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲,其与1-溴-2-取代乙酮环合,合成了糖基噻唑—N-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)-2-氨基-4-取代噻唑。本发明操作简单安全、适用范围广,原料廉价易得、后处理简便、收率高,是一种快速高效的合成方法,同时合成的化合物对乙酰胆碱酯酶有强的抑制作用,在制备抗乙酰胆碱酯酶药物方面,具有广阔的应用前景。
权利要求
1.一种氨糖噻唑衍生物,其特征在于,其结构式如下式Ⅶ:
其中,所述的R选自CH3-, n-C4H9-, C6H5-, 4-CH3C6H4-, 3-CH3C6H4-, 2-CH3C6H4-, 4-CH3OC6H4-, 3-CH3OC6H4-, 2,3-di-CH3OC6H3-, 4-FC6H4-, 2-FC6H4-, 4-ClC6H4-, 3-ClC6H4-,4-BrC6H4-, 3-BrC6H4-, 4-NO2C6H4-,3-NO2C6H4-, 4-OHC6H4-,4-NH2C6H4-, 4-PhC6H4-, 3-C6H4N-, 2-C5H3S-。
2.一种如权利要求1所述的氨糖噻唑衍生物的合成方法,其特征在于,其步骤如下:
(1)N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲制备:先将2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐与苯甲酰异硫氰酸酯作用,产物与碱反应得糖基硫脲;1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲与碱的摩尔比为1:1~2,反应温度为50~70℃,反应时间为0.5~1.5小时;
(2)N-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)-2-氨基-4-取代噻唑:1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲与1-溴-2-取代乙酮反应得到N-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)-2-氨基-4-取代噻唑;反应以乙醇为溶剂,N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲与1-溴-2-取代乙酮的摩尔比为1:1~1.5,反应温度为50~80℃,反应时间为0.2~0.5小时;1-溴-2-取代乙酮的取代基选自CH3-, n-C4H9-, C6H5-, 4-CH3C6H4-, 3-CH3C6H4-, 2-CH3C6H4-, 4-CH3OC6H4-, 3-CH3OC6H4-, 2,3-di-CH3OC6H3-, 4-FC6H4-, 2-FC6H4-, 4-ClC6H4-, 3-ClC6H4-,4-BrC6H4-, 3-BrC6H4-, 4-NO2C6H4-,3-NO2C6H4-, 4-OHC6H4-,4-NH2C6H4-, 4-PhC6H4-, 3-C6H4N-, 2-C5H3S-。
3.根据权利要求2所述的合成方法,其特征在于,步骤(1)所述的N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲的具体制备方法如下:在2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐的二氯甲烷溶液中,搅拌下滴加三乙胺;再与苯甲酰异硫氰酸酯反应得1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲;2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐与苯甲酰异硫氰酸酯的摩尔比为1:1~1.5,反应温度为10~30℃,反应时间为0.5~2小时;1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲在甲醇溶液中与碱反应得N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲。
4.根据权利要求2所述的合成方法,其特征在于,步骤(1)中原料2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐的制备方法如下:先将市售氨基葡萄糖盐酸盐与氢氧化钠反应得到氨基葡萄糖,再与对甲氧基苯甲醛反应得到对甲氧基苯甲醛缩-β-D-氨基葡萄糖稀夫碱;氨基葡萄糖盐酸盐与氢氧化钠摩尔比为1:1~1.5,反应温度为10~30℃,反应时间为0.5~2小时;氨基葡萄糖盐酸盐与对甲氧基苯甲醛的摩尔比为1:1~2,反应温度为10~30℃,反应时间为2~8小时;继而与溴化苄、氢化钠在N,N-二甲基甲酰胺溶剂下反应制得对甲氧基苯甲醛缩-β-D-氨基葡萄糖四苄基醚;希夫碱与溴化苄的摩尔比为1:4~10,反应温度为0~15℃,反应时间为5~10小时;希夫碱与氢化钠的摩尔比为1:4~10,反应温度为0~15℃,反应时间为5~10小时;最后在丙酮溶剂中与浓盐酸反应制得2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐,对甲氧基苯甲醛缩-β-D-氨基葡萄糖四苄基醚与浓盐酸的摩尔比为1:1.2~2,反应温度为50~80℃,反应时间为0.5~1小时。
5.根据权利要求2所述的合成方法,其特征在于:步骤(1)中,所述的碱为氢氧化钠、氢氧化钾、碳酸钾、碳酸钠、水合肼和甲醇钠。
6.根据权利要求2所述的合成方法,其特征在于:步骤(2)中,反应时间为0.3~0.4小时。
7.权利要求1所述的一种氨糖噻唑衍生物或者权利要求2-6中任何一项方法所合成的氨糖噻唑衍生物在制备治疗或者预防阿尔茨海默病药物中的应用。
说明书
技术领域
本发明涉及药物制备领域,具体涉及一种氨糖噻唑衍生物,本发明还涉及该氨糖噻唑衍生物的制备方法与用途。
背景技术
阿尔茨海默病(Alzheimer’s disease, AD)即老年痴呆症是一种常见的脑神经退行性病变,发病率较高,已成为现代社会严重威胁人类生命的疾病之一。其临床表现为认知和记忆功能不断恶化,日常生活能力进行性减退,并有各种神经精神症状和行为障碍。在发达国家,AD已成为继心脏病、癌症、中风之后导致老年人死亡的第4大杀手。在中国65岁以上的老人患病率高达6.6%以上,年龄每增加5岁,患病率增长一倍,3个85岁以上的老人中就有一个是老年痴呆。保守估计国内有老年痴呆患者500万,并仍以每年30万人的速度递增。此外,AD的治疗和护理费用昂贵,英国每年用于AD医护的费用为110亿美元,而美国更高达839亿美元。因此,AD的治疗已经不仅仅是一个医学问题,甚至已经成为一个亟待解决的严峻社会问题。
目前,AD发病确切的致病原因仍不太确定,临床上比较公认的是胆碱能假设。该假设认为,AD患者脑内缺乏一个重要的神经递质,乙酰胆碱,这种神经递质水平的减少引起了胆碱能神经传递的障碍从而导致认知和记忆功能的损伤。这个假设也就意味着增加大脑中乙酰胆碱的水平将能够改善或者治疗AD患者的认知功能。临床试验表明,乙酰胆碱酯酶抑制剂(acetylcholinesterase, AChE)能够抑制AChE的活性,从而降低或延缓乙酰胆碱的水解速度,恢复突触间隙乙酰胆碱的水平,从而达到对AD的控制与治疗的功效。
虽然现有的AChE抑制剂已经成为治疗AD的主流药物,但是仅仅抑制乙酰胆碱酯酶的活性只能提高乙酰胆碱的含量,不能阻止中枢胆碱能神经元的进行性退化死亡。随着病情的发展,中枢胆碱能神经元发生进行性退化死亡,乙酰胆碱酯酶抑制剂的药效也会逐渐降低。并且现有的AChE抑制剂普遍存在着药物选择性差、生物利用度不高、胃肠刺激性大副作用、治疗范围窄以及严重的肝细胞毒性等缺点。因此制备和发现结构新颖,活性更强且不良反应小的乙酰胆碱酯酶抑制剂,对于治疗老年性痴呆病具有十分重要意义。
发明内容
本发明所要解决的技术问题是针对现有技术的不足,提供一种新的糖基噻唑衍生物。
本发明所要解决的另一个技术问题是提供了一种快速,高效,高产合成糖基噻唑衍生物的方法。
本发明所要解决的再一技术问题是提供上述糖基噻唑衍生物在乙酰胆碱酯酶抑制方面的应用。
本发明所要解决的技术问题是通过以下技术方案来实现的。本发明是一种氨糖噻唑衍生物,其结构式如式(Ⅶ)所示:
其中,所述的R选自CH3-, n-C4H9-, C6H5-, 4-CH3C6H4-, 3-CH3C6H4-, 2-CH3C6H4-, 4-CH3OC6H4-, 3-CH3OC6H4-, 2,3-di-CH3OC6H3-, 4-FC6H4-, 2-FC6H4-, 4-ClC6H4-, 3-ClC6H4-,4-BrC6H4-, 3-BrC6H4-, 4-NO2C6H4-,3-NO2C6H4-, 4-OHC6H4-,4-NH2C6H4-, 4-PhC6H4-, 3-C6H4N-, 2-C5H3S-。
本发明所要解决的技术问题还可以通过以下的技术方案来实现。本发明还公开了一种如以上技术方案所述的氨糖噻唑衍生物的合成方法,其特点是,其步骤如下:
(1)N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲制备:先将2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐与苯甲酰异硫氰酸酯作用,产物与碱反应得糖基硫脲;1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲与碱的摩尔比为1:1~2,反应温度为50~70℃,反应时间为0.5~1.5小时;
(2)N-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)-2-氨基-4-取代噻唑:1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲与1-溴-2-取代乙酮反应得到N-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)-2-氨基-4-取代噻唑;反应以乙醇为溶剂,N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲与1-溴-2-取代乙酮的摩尔比为1:1~1.5,反应温度为50~80℃,反应时间为0.2~0.5小时;1-溴-2-取代乙酮的取代基选自CH3-, n-C4H9-, C6H5-, 4-CH3C6H4-, 3-CH3C6H4-, 2-CH3C6H4-, 4-CH3OC6H4-, 3-CH3OC6H4-, 2,3-di-CH3OC6H3-, 4-FC6H4-, 2-FC6H4-, 4-ClC6H4-, 3-ClC6H4-,4-BrC6H4-, 3-BrC6H4-, 4-NO2C6H4-,3-NO2C6H4-, 4-OHC6H4-,4-NH2C6H4-, 4-PhC6H4-, 3-C6H4N-, 2-C5H3S-。
本发明所述的合成方法的步骤(1)所述的N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲的优选的制备方法如下:在2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐的二氯甲烷溶液中,搅拌下滴加三乙胺;再与苯甲酰异硫氰酸酯反应得1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲;2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐与苯甲酰异硫氰酸酯的摩尔比为1:1~1.5,反应温度为10~30℃,反应时间为0.5~2小时;1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲在甲醇溶液中与碱反应得N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲。
本发明所述的合成方法技术方案中,各原料均可以采用市售或以其它方式公开的产品。其中,步骤(1)中原料2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐的优选的制备方法如下:先将市售氨基葡萄糖盐酸盐与氢氧化钠反应得到氨基葡萄糖,再与对甲氧基苯甲醛反应得到对甲氧基苯甲醛缩-β-D-氨基葡萄糖稀夫碱;氨基葡萄糖盐酸盐与氢氧化钠摩尔比为1:1~1.5,反应温度为10~30℃,反应时间为0.5~2小时;氨基葡萄糖盐酸盐与对甲氧基苯甲醛的摩尔比为1:1~2,反应温度为10~30℃,反应时间为2~8小时;继而与溴化苄、氢化钠在N,N-二甲基甲酰胺溶剂下反应制得对甲氧基苯甲醛缩-β-D-氨基葡萄糖四苄基醚;希夫碱与溴化苄的摩尔比为1:4~10,反应温度为0~15℃,反应时间为5~10小时;希夫碱与氢化钠的摩尔比为1:4~10,反应温度为0~15℃,反应时间为5~10小时;最后在丙酮溶剂中与浓盐酸反应制得2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐,对甲氧基苯甲醛缩-β-D-氨基葡萄糖四苄基醚与浓盐酸的摩尔比为1:1.2~2,反应温度为50~80℃,反应时间为0.5~1小时。
本发明所述的合成方法的步骤(1)中,所述的碱可以为常规碱,优选为氢氧化钠、氢氧化钾、碳酸钾、碳酸钠、水合肼和甲醇钠。
本发明所述的合成方法的步骤(2)中,反应时间优选为0.3~0.4小时。
本发明所述的一种氨糖噻唑衍生物可以在制备治疗或者预防阿尔茨海默病药物中得到应用。
本发明合成方法的路线如下:
本发明合成方法的步骤中,反应可以在传统加热方式的条件下进行。传统加热方式有燃气灯,电热套,水浴,油浴,水蒸气浴,沙浴,盐浴,金属浴等等。
本发明合成方法的优点是:苄基保护的糖基噻唑是一种快速,高效,高收率的合成方法。本方法以市售氨基葡萄糖盐酸盐、溴化苄、苯甲酰异硫氰酸酯和1-溴-2-取代乙酮为原料,原料廉价易得,降低了成本;反应条件采用传统加热模式,实验路线简捷,后处理简便环保,产品收率高,拓宽了该方法的适用范围;采用廉价易得原料作为反应物,降低了生产成本;在合成的目标化合物中引入噻唑环结构,有望得到生物活性更高的物质;合成的目标化合物对乙酰胆碱酯酶有强的抑制活性,最强的活性其IC50达到了0.54μM,与传统治疗老年痴呆药物他克林活性约0.2μM具有可比性,因此在制备抗乙酰胆碱酯酶药物方面,具有广阔的应用前景。
本发明氨糖噻唑衍生物鉴定采用了红外光谱、氢谱和高分辨率质谱:红外光谱采用布鲁克FT-IR-TENSOR-27光谱仪,溴化钾压片;氢谱采用布鲁克超导核磁共振仪(Bruker ACF 400 MHz spectrometer),常用四甲基硅烷作为标准物质,DMSO-d6做溶剂;质谱采用安捷伦高分辨质谱仪(Agilent 6230 TOF spectrometer)。
红外光谱图中:在3360~3450 cm-1处出现吸收峰,为分子中仲胺的伸缩振动吸收峰;在2850~2950 cm-1处出现吸收峰,为苄基中亚甲基的伸缩振动吸收峰;在1610~1650 cm-1处出现吸收峰,为噻唑环上碳氮双键伸缩振动吸收峰;在1450~1550 cm-1处出现吸收峰,为苯环的骨架振动吸收峰。
氢谱图中:δ值在7.90~8.2 ppm处的双峰为结构中仲胺N-H的吸收峰,7.83-7.11 ppm之间出现的峰是苯环上质子的吸收峰,7.05~7.5 ppm处的单峰为噻唑环上C=CH中质子吸收峰,4.83-4.57 ppm出现的峰是苄基上亚甲基中的氢和糖环氢吸收峰,3.80-3.90 ppm处峰的是糖环头碳质子吸收峰,其耦合常数为9.1 Hz,说明化合物为β构型。
质谱图中:在高分辨质谱图中均能显示其分子离子峰。
从红外光谱图、核磁共振氢谱和高分辨质谱图可以证明,本发明合成方法得到的苄基保护的糖基噻唑与理论产物结构一致。
具体实施方式
以下进一步描述发明的具体技术方案,以便于本领域的技术人员进一步地理解本发明,而不构成对其权利的限制。
实施例1,一种氨糖噻唑衍生物,其结构式如下式Ⅶ:
其中,所述的R选自CH3-, n-C4H9-, C6H5-, 4-CH3C6H4-, 3-CH3C6H4-, 2-CH3C6H4-, 4-CH3OC6H4-, 3-CH3OC6H4-, 2,3-di-CH3OC6H3-, 4-FC6H4-, 2-FC6H4-, 4-ClC6H4-, 3-ClC6H4-,4-BrC6H4-, 3-BrC6H4-, 4-NO2C6H4-,3-NO2C6H4-, 4-OHC6H4-,4-NH2C6H4-, 4-PhC6H4-, 3-C6H4N-, 2-C5H3S-。
实施例2,一种如实施例1所述的氨糖噻唑衍生物的合成方法,其步骤如下:
(1)N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲制备:先将2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐与苯甲酰异硫氰酸酯作用,产物与碱反应得糖基硫脲;1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲与碱的摩尔比为1:1,反应温度为50℃,反应时间为0.5小时;
(2)N-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)-2-氨基-4-取代噻唑:1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲与1-溴-2-取代乙酮反应得到N-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)-2-氨基-4-取代噻唑;反应以乙醇为溶剂,N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲与1-溴-2-取代乙酮的摩尔比为1:1,反应温度为50℃,反应时间为0.2小时;1-溴-2-取代乙酮的取代基选自CH3-, n-C4H9-, C6H5-, 4-CH3C6H4-, 3-CH3C6H4-, 2-CH3C6H4-, 4-CH3OC6H4-, 3-CH3OC6H4-, 2,3-di-CH3OC6H3-, 4-FC6H4-, 2-FC6H4-, 4-ClC6H4-, 3-ClC6H4-,4-BrC6H4-, 3-BrC6H4-, 4-NO2C6H4-,3-NO2C6H4-, 4-OHC6H4-,4-NH2C6H4-, 4-PhC6H4-, 3-C6H4N-, 2-C5H3S-。
实施例3,一种如实施例1所述的氨糖噻唑衍生物的合成方法,其步骤如下:
(1)N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲制备:先将2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐与苯甲酰异硫氰酸酯作用,产物与碱反应得糖基硫脲;1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲与碱的摩尔比为1: 2,反应温度为70℃,反应时间为1.5小时;
(2)N-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)-2-氨基-4-取代噻唑:1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲与1-溴-2-取代乙酮反应得到N-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)-2-氨基-4-取代噻唑;反应以乙醇为溶剂,N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲与1-溴-2-取代乙酮的摩尔比为1: 1.5,反应温度为80℃,反应时间为0.5小时;1-溴-2-取代乙酮的取代基选自CH3-, n-C4H9-, C6H5-, 4-CH3C6H4-, 3-CH3C6H4-, 2-CH3C6H4-, 4-CH3OC6H4-, 3-CH3OC6H4-, 2,3-di-CH3OC6H3-, 4-FC6H4-, 2-FC6H4-, 4-ClC6H4-, 3-ClC6H4-,4-BrC6H4-, 3-BrC6H4-, 4-NO2C6H4-,3-NO2C6H4-, 4-OHC6H4-,4-NH2C6H4-, 4-PhC6H4-, 3-C6H4N-, 2-C5H3S-。
实施例4,一种如实施例1所述的氨糖噻唑衍生物的合成方法,其步骤如下:
(1)N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲制备:先将2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐与苯甲酰异硫氰酸酯作用,产物与碱反应得糖基硫脲;1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲与碱的摩尔比为1:1.2,反应温度为60℃,反应时间为1小时;
(2)N-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)-2-氨基-4-取代噻唑:1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲与1-溴-2-取代乙酮反应得到N-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)-2-氨基-4-取代噻唑;反应以乙醇为溶剂,N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲与1-溴-2-取代乙酮的摩尔比为1: 1.5,反应温度为65℃,反应时间为0.3小时;1-溴-2-取代乙酮的取代基选自CH3-, n-C4H9-, C6H5-, 4-CH3C6H4-, 3-CH3C6H4-, 2-CH3C6H4-, 4-CH3OC6H4-, 3-CH3OC6H4-, 2,3-di-CH3OC6H3-, 4-FC6H4-, 2-FC6H4-, 4-ClC6H4-, 3-ClC6H4-,4-BrC6H4-, 3-BrC6H4-, 4-NO2C6H4-,3-NO2C6H4-, 4-OHC6H4-,4-NH2C6H4-, 4-PhC6H4-, 3-C6H4N-, 2-C5H3S-。
实施例5,实施例2-4所述的氨糖噻唑衍生物的合成方法中:步骤(1)所述的N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲的具体制备方法如下:在2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐的二氯甲烷溶液中,搅拌下滴加三乙胺;再与苯甲酰异硫氰酸酯反应得1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲;2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐与苯甲酰异硫氰酸酯的摩尔比为1:1~1.5,反应温度为10~30℃,反应时间为0.5~2小时;1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲在甲醇溶液中与碱反应得N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲。
实施例6,实施例2-4所述的氨糖噻唑衍生物的合成方法中:步骤(1)所述的N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲的具体制备方法如下:在2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐的二氯甲烷溶液中,搅拌下滴加三乙胺;再与苯甲酰异硫氰酸酯反应得1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲;2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐与苯甲酰异硫氰酸酯的摩尔比为1: 1.5,反应温度为20℃,反应时间为1小时;1-苯甲酰基-3-(1,3,4,6-四-O-苄基-β-D-吡喃葡萄糖-2-基)硫脲在甲醇溶液中与碱反应得N-(1,3,4,6-四-O-苄基-2-脱氧-β-D-吡喃葡萄糖-2-基)硫脲。
实施例7,实施例2-6所述的氨糖噻唑衍生物的合成方法中:步骤(1)中原料2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐的制备方法如下:先将市售氨基葡萄糖盐酸盐与氢氧化钠反应得到氨基葡萄糖,再与对甲氧基苯甲醛反应得到对甲氧基苯甲醛缩-β-D-氨基葡萄糖稀夫碱;氨基葡萄糖盐酸盐与氢氧化钠摩尔比为1:1~1.5,反应温度为10~30℃,反应时间为0.5~2小时;氨基葡萄糖盐酸盐与对甲氧基苯甲醛的摩尔比为1:1~2,反应温度为10~30℃,反应时间为2~8小时;继而与溴化苄、氢化钠在N,N-二甲基甲酰胺溶剂下反应制得对甲氧基苯甲醛缩-β-D-氨基葡萄糖四苄基醚;希夫碱与溴化苄的摩尔比为1:4~10,反应温度为0~15℃,反应时间为5~10小时;希夫碱与氢化钠的摩尔比为1:4~10,反应温度为0~15℃,反应时间为5~10小时;最后在丙酮溶剂中与浓盐酸反应制得2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐,对甲氧基苯甲醛缩-β-D-氨基葡萄糖四苄基醚与浓盐酸的摩尔比为1:1.2~2,反应温度为50~80℃,反应时间为0.5~1小时。
实施例8,实施例2-6所述的氨糖噻唑衍生物的合成方法中:步骤(1)中原料2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐的制备方法如下:先将市售氨基葡萄糖盐酸盐与氢氧化钠反应得到氨基葡萄糖,再与对甲氧基苯甲醛反应得到对甲氧基苯甲醛缩-β-D-氨基葡萄糖稀夫碱;氨基葡萄糖盐酸盐与氢氧化钠摩尔比为1: 1.5,反应温度为20℃,反应时间为1小时;氨基葡萄糖盐酸盐与对甲氧基苯甲醛的摩尔比为1:1.5,反应温度为20℃,反应时间为6小时;继而与溴化苄、氢化钠在N,N-二甲基甲酰胺溶剂下反应制得对甲氧基苯甲醛缩-β-D-氨基葡萄糖四苄基醚;希夫碱与溴化苄的摩尔比为1:7,反应温度为10℃,反应时间为7小时;希夫碱与氢化钠的摩尔比为1:7,反应温度为10℃,反应时间为7小时;最后在丙酮溶剂中与浓盐酸反应制得2-脱氧-2-氨基-1,3,4,6-四-O-苄基-β-D-吡喃糖盐酸盐,对甲氧基苯甲醛缩-β-D-氨基葡萄糖四苄基醚与浓盐酸的摩尔比为1:1.2,反应温度为65℃,反应时间为0.5~1小时。
实施例9,实施例2-8所述的氨糖噻唑衍生物的合成方法中:步骤(1)中,所述的碱为氢氧化钠、氢氧化钾、碳酸钾、碳酸钠、水合肼和甲醇钠。
实施例10,一种如实施例1所述的氨糖噻唑衍生物的合成方法,其步骤如下:
(1)化合物Ⅳ(化合物的代号与前述的反应路线中的一致,下同)的制备:化合物Ⅰ先与氢氧化钠反应,再与对甲氧基苯甲醛作用,得化合物Ⅱ,化合物Ⅰ与氢氧化钠的摩尔比为1:1.5,反应温度30℃,反应时间0.5小时;化合物Ⅰ与对甲氧基苯甲醛的摩尔比为1:2,反应温度30℃,反应时间2小时;化合物Ⅱ在冰水浴条件下与溴化苄、氢化钠作用,合成化合物Ⅲ,化合物Ⅱ与溴化苄、氢化钠的摩尔比分别为1:4,1:4,反应温度为15℃,反应时间5小时;化合物Ⅲ与盐酸在丙酮中反应,合成化合物Ⅳ,化合物Ⅲ与盐酸的摩尔比为1:2.5,反应温度为50℃,反应时间1小时;
(2)化合物Ⅵ的制备:化合物Ⅳ与苯甲酰异硫氰酸酯在二氯甲烷中反应,得到化合物Ⅴ,化合物Ⅳ与苯甲酰异硫氰酸酯的摩尔比为1:1.5,反应温度30℃,反应时间2小时;化合物Ⅴ与碱在甲醇溶液中反应,得到化合物Ⅵ,化合物Ⅴ与碱的摩尔比为1:2,反应温度50℃,反应时间1.5小时;
(3)化合物Ⅶ的制备:化合物Ⅵ与1-溴-2-取代乙酮在乙醇中反应得化合物Ⅶ,化合物Ⅵ与1-溴-2-取代乙酮的摩尔比为1:1.5,反应温度50℃,反应时间0.5小时。
实施例1,一种如实施例1所述的氨糖噻唑衍生物的合成方法,其步骤如下:
(1)化合物Ⅳ的制备:化合物Ⅰ先与氢氧化钠反应,再与对甲氧基苯甲醛作用,得化合物Ⅱ,化合物Ⅰ与氢氧化钠的摩尔比为1:1.3,反应温度20℃,反应时间1小时;化合物Ⅰ与对甲氧基苯甲醛的摩尔比为1:1.8,反应温度20℃,反应时间4小时;化合物Ⅱ在冰水浴条件下与溴化苄、氢化钠作用,合成化合物Ⅲ,化合物Ⅱ与溴化苄、氢化钠的摩尔比分别为1:6,1:6,反应温度为10℃,反应时间7小时;化合物Ⅲ与盐酸在丙酮中反应,合成化合物Ⅳ,化合物Ⅲ与盐酸的摩尔比为1:2,反应温度为60℃,反应时间1小时;
(2)化合物Ⅵ的制备:化合物Ⅳ与苯甲酰异硫氰酸酯在二氯甲烷中反应,得到化合物Ⅴ,化合物Ⅳ与苯甲酰异硫氰酸酯的摩尔比为1:1.3,反应温度20℃,反应时间1.5小时;化合物Ⅴ与碱在甲醇溶液中反应,得到化合物Ⅵ,化合物Ⅴ与碱的摩尔比为1:1.5,反应温度60℃,反应时间1小时;
(3)化合物Ⅶ的制备:化合物Ⅵ与1-溴-2-取代乙酮在乙醇中反应得化合物Ⅶ,化合物Ⅵ与1-溴-2-取代乙酮的摩尔比为1:1.3,反应温度60℃,反应时间0.4小时。
实施例12,一种如实施例1所述的氨糖噻唑衍生物的合成方法,其步骤如下:
(1)化合物Ⅳ的制备:化合物Ⅰ先与氢氧化钠反应,再与对甲氧基苯甲醛作用,得化合物Ⅱ,化合物Ⅰ与氢氧化钠的摩尔比为1:1.2,反应温度10℃,反应时间1小时;化合物Ⅰ与对甲氧基苯甲醛的摩尔比为1:1.6,反应温度20℃,反应时间4小时;化合物Ⅱ在冰水浴条件下与溴化苄、氢化钠作用,合成化合物Ⅲ,化合物Ⅱ与溴化苄、氢化钠的摩尔比分别为1:8,1:8,反应温度为5℃,反应时间9小时;化合物Ⅲ与盐酸在丙酮中反应,合成化合物Ⅳ,化合物Ⅲ与盐酸的摩尔比为1:1.5,反应温度为70℃,反应时间1小时;
(2)化合物Ⅵ的制备:化合物Ⅳ与苯甲酰异硫氰酸酯在二氯甲烷中反应,得到化合物Ⅴ,化合物Ⅳ与苯甲酰异硫氰酸酯的摩尔比为1:1.2,反应温度30℃,反应时间2小时;化合物Ⅴ与碱在甲醇溶液反应,得到化合物Ⅵ,化合物Ⅴ与碱的摩尔比为1:2,反应温度70℃,反应时间1小时;
(3)化合物Ⅶ的制备:糖基硫脲(Ⅵ)与1-溴-2-取代乙酮在乙醇中反应得化合物Ⅶ。化合物Ⅵ与1-溴-2-取代乙酮的摩尔比为1:1.2,反应温度70℃,反应时间0.3小时。
实施例13,一种如实施例1所述的氨糖噻唑衍生物的合成方法,其步骤如下:
(1)化合物Ⅳ的制备:化合物Ⅰ先与氢氧化钠反应,再与对甲氧基苯甲醛作用,得化合物Ⅱ,化合物Ⅰ与氢氧化钠的摩尔比为1:1.1,反应温度10℃,反应时间0.5小时;化合物Ⅰ与对甲氧基苯甲醛的摩尔比为1:1.3,反应温度10℃,反应时间6小时;化合物Ⅱ在冰水浴条件下与溴化苄、氢化钠作用,合成化合物Ⅲ,化合物Ⅱ与溴化苄、氢化钠的摩尔比分别为1:9,1:9,反应温度为0℃,反应时间10小时;化合物Ⅲ与盐酸在丙酮中反应,合成化合物Ⅳ,化合物Ⅲ与盐酸的摩尔比为1:1.2,反应温度为80℃,反应时间1小时;
(2)化合物Ⅵ的制备:化合物Ⅳ与苯甲酰异硫氰酸酯在二氯甲烷中反应,得到化合物Ⅴ,化合物Ⅳ与苯甲酰异硫氰酸酯的摩尔比为1:1.1,反应温度30℃,反应时间2小时;化合物Ⅴ与碱在甲醇溶液中反应得到化合物Ⅵ,化合物Ⅴ与碱的摩尔比为1:1.5,反应温度70℃,反应时间1.5小时;
(3)化合物Ⅶ的制备:化合物Ⅵ与1-溴-2-取代乙酮在乙醇中反应得化合物Ⅶ,化合物Ⅵ与1-溴-2-取代乙酮的摩尔比为1:1,反应温度80℃,反应时间0.3小时。
实施例14,一种如实施例1所述的氨糖噻唑衍生物的合成方法,其步骤如下:
(1)化合物Ⅳ的制备:化合物Ⅰ先与氢氧化钠反应,再与对甲氧基苯甲醛作用,得化合物Ⅱ,化合物Ⅰ与氢氧化钠的摩尔比为1:1,反应温度10℃,反应时间0.5小时;化合物Ⅰ与对甲氧基苯甲醛的摩尔比为1:1,反应温度10℃,反应时间8小时;化合物Ⅱ在冰水浴条件下与溴化苄、氢化钠作用,合成化合物Ⅲ,化合物Ⅱ与溴化苄、氢化钠的摩尔比分别为1:10,1:10,反应温度为0℃,反应时间10小时;化合物Ⅲ与盐酸在丙酮中反应,合成化合物Ⅳ,化合物Ⅲ与盐酸的摩尔比为1:1.2,反应温度为80℃,反应时间0.5小时;
(2)化合物Ⅵ的制备:化合物Ⅳ与苯甲酰异硫氰酸酯在二氯甲烷中反应,得到化合物Ⅴ,化合物Ⅳ与苯甲酰异硫氰酸酯的摩尔比为1:1,反应温度30℃,反应时间0.5小时;化合物Ⅴ与碱在甲醇溶液中反应得到化合物Ⅵ,化合物Ⅴ与碱的摩尔比为1:1,反应温度70℃,反应时间0.5小时;
(3)化合物Ⅶ的制备:化合物Ⅵ与1-溴-2-取代乙酮在乙醇中反应得化合物Ⅶ,化合物Ⅵ与1-溴-2-取代乙酮的摩尔比为1:1,反应温度80℃,反应时间0.2小时。
上述实施例10-14中所述的合成方法在传统加热方式的条件下进行。化合物Ⅵ的制备中所用的碱为氢氧化钠、氢氧化钾、碳酸钾、碳酸钠、水合肼和甲醇钠。
实施例15,一种如实施例1所述的氨糖噻唑衍生物的合成方法实验:
(1)实验方法参照实施例13;
(2)制备化合物Ⅶ的反应条件、产物收率和取代基结构如下表所示:
(3)对制得的化合物进行乙酰胆碱胆碱酯酶抑制实验:
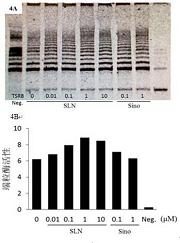
采用碘化乙酰胆碱(ATCI)为底物,二硫硝基苯甲酸(DTNB)为显色剂,在96孔板上测定样品对AChE的抑制活性。在96孔板中加入样品10 μL(甲醇溶解),AChE酶液20 μL,使用Synergy HT全自动酶标仪,在410 nm波长下,读取每孔光密度OD值(样品本底OD值),25 ℃保温20 min后,加入底物与显色剂以及磷酸缓冲液至总体积为200 μL,底物与显色剂终浓度分别为0.5 μmol/L和0.25 mmol/L,20 ℃保温20 min后,读取每孔的OD值,并记录数据。用如下公式来计算酶活性:
抑制率(%)=[(B-S) / B] ×100%;
其中B为加空白甲醇时的吸光度变化值,S为样品的吸光度变化值。测定5 个浓度的样品,绘制剂量-抑制率曲线,计算其IC50 值。每个样品重复测定三次,结果用平均值±标准偏差表示。以他克林为阳性对照药。
[0029] 以酶的相对活力对抑制剂浓度作图,根据抑制曲线求得各种化合物的IC50值(抑制酶活力50% 时的抑制剂浓度)。
结果测得合成的目标化合物氨糖噻唑衍生物对乙酰胆碱酯酶均有不同程度的抑制作用,其中最佳IC50 = 0.54±0.18 μM。
综上可知,本发明操作简单安全、适用范围广,原料廉价易得、后处理简便、收率高,是一种快速高效的合成方法,同时合成的化合物对乙酰胆碱酯酶均有不同程度的抑制作用,因而此类结构化合物可在制备抗乙酰胆碱酯酶药物方面具有广阔的应用前景,可以用来制备治疗或者预防阿尔茨海默病的药物。
一种氨糖噻唑衍生物及其合成方法与用途专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。









动态评分
0.0