







专利摘要
本发明提供了一种提纯喜果苷的方法,属于天然植物单体提纯技术领域。该方法首先以乙醇水溶液提取喜树果,得到喜树果粗提物,再利用三带模拟移动床色谱分离提纯喜树果粗提物中的喜果苷,得到喜果苷萃取液,再将萃取液进行浓缩和结晶后处理得到高纯度的喜果苷。该方法可以规模化、高效地提纯喜果苷,并且模拟移动床色谱分离过程实现了稳健、连续、自动运行,固定相和流动相能反复利用,降低了提纯成本。
权利要求
1.一种提纯喜果苷的方法,其特征在于,包括如下步骤:
(1)制备喜树果粗提物
喜树果原料粉粹后,用溶剂浸取1-3次,得到浸取液;将浸取液浓缩后,加乙醇或水和乙醇,使该混合液体积为所述浸取液体积的1/5~1/20,并且该混合液中乙醇的体积百分含量为60~100%;除去混合液中产生的沉淀,得滤液;再用水和乙醇稀释滤液,得喜树果粗提物进样液;
喜树果原料和溶剂的料液比为1g∶10mL~1g∶20mL;所述溶剂中乙醇的体积百分含量为50~100%,余量为水;
喜树果粗提物进样液中喜果苷的浓度为0.1~3mg/mL,乙醇的体积百分含量为40~65%;
(2)用模拟移动床色谱简称SMBC提纯喜果苷
1)SMBC分离条件如下:
色谱柱:3~10根;
固定相:十八烷基硅烷键合硅胶ODS,10~60μm;
运行模式:设置三带SMBC,三带包括洗脱带、精制带和吸附带,a为洗脱带色谱柱数目,1≤a≤2;b为精制带色谱柱数目,1≤b≤4;c为吸附带色谱柱数目,1≤c≤4,运行模式用a-b-c表示;
流动相组成:洗脱带的流动相P为乙醇与水的混合溶液或乙醇,流动相P中乙醇的体积百分含量为50~100%;精制带和吸附带的流动相D为乙醇与水的混合溶液,乙醇的体积百分含量为50~70%;
流动相流速:在精制带和吸附带中,洗脱液D流速Q
切换时间T
模拟移动床操作温度:室温;
2)SMBC分离过程如下:
在上述SMBC工作条件下进行SMBC分离,由原料液入口连续泵入喜树果粗提物进样液,SMBC自动控制系统按切换时间T
(3)对萃取液进行后处理
将喜果苷萃取液浓缩至浑浊,静置分层,弃去上层乳白色液体,用蒸馏水对下层沉淀进行清洗,直至清洗液无色;再用乙醇对下层沉淀重结晶,得到喜果苷产品。
2.根据权利要求1所述的一种提纯喜果苷的方法,其特征在于,由甲醇替代乙醇作为溶剂或流动相。
说明书
技术领域
本发明属于天然植物单体提纯技术领域,特别涉及一种提纯喜果苷的方法。
背景技术
喜树果是我国特有树种喜树的果实,自古以来,喜树在民间即为重要药材,且可全株用药,用于治疗恶性疗毒,肿瘤。喜树果中的主要化学成分包括:喜树碱、10-羟基喜树碱、11-甲氧基喜树碱、脱氧喜树碱、喜树次碱、白桦酯酸及喜果苷。研究发现,喜果苷和喜树碱类化合物均有抗癌活性,喜果苷还具有抗菌,抗病毒,抗炎,阵痛,解热的用途。
由喜树果中提取喜果苷,具有重要实用价值。目前提取分离喜果苷的方法主要是柱色谱法,如:欧来良等人(CN 1295239C,2007)将喜树果用乙醇溶液渗漉或乙醇溶液25-80℃热提取、提取液通入装有大孔吸附树脂(AL-2吸附树脂)的树脂柱中吸附、用乙醇水溶液洗涤和解吸、流出液旋蒸浓缩和重结晶等步骤得喜果苷产品;王瑞芳等人(应用化学,2005,22(1):24-29)用合成的大孔超高交联吸附树脂(Rf18)及其甲胺化(Rs3)、N-甲基乙酰胺化(Rs4)、二甲胺化(Rs5)、三甲胺化(Rs6)后对喜树碱和喜果苷进行柱层析分离;史伟国等人(中国中药杂志,2008,32(21):2487-2489)用聚酰胺分离纯化10-羟基喜树碱和喜果苷;王瑞芳等人(应用化学,2009,26(5):593-596)通过选用不同结构的纳米吸附树脂对喜树果提取液中的喜树碱和喜果苷(CPT和VCS-LT)进行层析分离。现有柱色谱法提纯喜果苷的工艺均为批处理工艺,洗脱剂耗量大,不能连续自动生产。
模拟移动床色谱(简称SMBC)是提纯化合物的高效分离手段。模拟移动床色谱在制备色谱的基础上,引进连续运行机制,能够进行自动化和规模化的分离。
发明内容
本发明的目的是针对现有技术中分离纯化喜果苷方法存在的不足,提供了一种提纯喜果苷的方法。该方法首先以乙醇水溶液提取喜树果粗提物,再利用三带模拟移动床色谱分离提纯喜树果粗提物中的喜果苷,得到喜果苷萃取液,再将萃取液进行浓缩和结晶后处理得到高纯度的喜果苷。该方法可以规模化、高效地提纯喜果苷,并且模拟移动床色谱分离过程实现了稳健、连续、自动运行,固定相和流动相能反复利用,降低了提纯成本。
一种提纯喜果苷的方法,包括如下步骤:
(1)制备喜树果粗提物,即:原料前处理
喜树果原料粉粹后,用溶剂浸取1-3次,得到浸取液;将浸取液浓缩后,加乙醇或水和乙醇,使该混合液体积为所述浸取液体积的1/5~1/20,并且该混合液中乙醇的体积百分含量为60~100%,除去混合液中产生的沉淀,得滤液;再用水和乙醇稀释滤液,得喜树果粗提物进样液;
所述喜树果原料和溶剂的料液比为1g∶10mL~1g∶20mL;所述溶剂中乙醇的体积百分含量为50~100%,余量为水;
所述喜树果粗提物进样液中喜果苷的浓度为0.1~3mg/mL,乙醇的体积百分含量为40~65%;
(2)用SMBC提纯喜果苷
1)SMBC分离条件如下:
色谱柱:3~10根;
固定相:十八烷基硅烷键合硅胶ODS,10~60μm;
运行模式:设置三带SMBC,三带包括洗脱带、精制带和吸附带,a为洗脱带色谱柱数目,1≤a≤2;b为精制带色谱柱数目,1≤b≤4;c为吸附带色谱柱数目,1≤c≤4,运行模式用a-b-c表示;
流动相组成:洗脱带的流动相P为乙醇与水的混合溶液或乙醇,流动相P中乙醇的体积百分含量为50~100%;精制带和吸附带的流动相D为乙醇与水的混合溶液,乙醇的体积百分含量为50~70%;
流动相流速:在精制带和吸附带中,洗脱液D流速QD为每小时1~10倍柱体积,即1~10BV/h,当洗脱液D流速QD单位为mL/min,色谱柱半径r单位为cm,色谱柱柱长L单位为cm时,则1个BV/h相当于60QD/(πr
切换时间TS:8~20min;
模拟移动床操作温度:室温;
2)SMBC分离过程如下:
在上述SMBC工作条件下进行SMBC分离,由原料液入口连续泵入喜树果粗提物进样液,SMBC自动控制系统按切换时间TS沿流动相方向将洗脱液入口、萃取液出口、原料液入口和萃余液出口的位置同时依次移动至下一根色谱柱,从萃余液R出口除去各种前杂质,从萃取液E出口得到喜果苷萃取液;
(3)对萃取液进行后处理
将含喜果苷萃取液浓缩至浑浊,静置分层,弃去上层乳白色液体,用蒸馏水对下层沉淀进行清洗,直至清洗液无色;再用乙醇对下层沉淀重结晶,得到喜果苷产品。
上述的一种提纯喜果苷的方法,可以用甲醇替代乙醇作为溶剂或流动相。
本发明与现有制备色谱分离技术相比,其显著的有益效果体现在:
1、本发明中乙醇-水溶液对喜树果原料的前处理能充分去除进样液不溶物,保证SMBC的稳定运行,由SMBC得到的萃取液再结合后处理,能够得到高纯度的喜果苷,纯度高于98%。
2、本发明利用SMBC技术能够规模化、稳健、连续、自动、高效地从喜树果中提纯喜果苷,在SMBC的萃取液中,产品回收率高于99%,HPLC纯度高于90%,固定相和流动相能反复利用,降低了提纯成本,属于绿色环保分离过程。
附图说明
图1、实施例1中喜树果进样液的HPLC谱图。
图2、实施例1中的三带SMBC体系。
图3、实施例1中三带SMBC萃余液的HPLC谱图。
图4、实施例1中三带SMBC萃取液的HPLC谱图。
图5、实施例1中经后处理的喜果苷产品的HPLC谱图。
图6、实施例1中经后处理的喜果苷产品的晶体扫描电镜图。
具体实施方式
一种提纯喜果苷的方法,包括如下步骤:
(1)制备喜树果粗提物
喜树果原料粉粹后,用溶剂浸取1-3次,得到浸取液;将浸取液浓缩后,加乙醇或水和乙醇,使该混合液体积为所述浸取液体积的1/5~1/20,并且该混合液中乙醇的体积百分含量为60~100%,除去混合液中产生的沉淀,得滤液;再用水和乙醇稀释滤液,得喜树果粗提物进样液;
所述喜树果原料和溶剂的料液比为1g∶10mL~1g∶20mL,溶剂中乙醇的体积百分含量为50~100%,余量为水;
所述喜树果粗提物进样液中喜果苷的浓度为0.1~3mg/mL,乙醇的体积百分含量为40~65%;
(2)用SMBC提纯喜果苷
1)SMBC分离条件如下:
色谱柱:3~10根;
固定相:十八烷基硅烷键合硅胶ODS,10~60μm;
运行模式:设置三带SMBC,三带包括洗脱带、精制带和吸附带,a为洗脱带色谱柱数目,1≤a≤2;b为精制带色谱柱数目,1≤b≤4;c为吸附带色谱柱数目,1≤c≤4,运行模式用a-b-c表示;
流动相组成:洗脱带的流动相P为乙醇与水的混合溶液或乙醇,流动相P中乙醇的体积百分含量为50~100%;精制带和吸附带的流动相D为乙醇与水的混合溶液,乙醇的体积百分含量为50~70%;
流动相流速:在精制带和吸附带中,洗脱液D流速QD为每小时1~10倍柱体积,即1~10BV/h,当洗脱液D流速QD单位为mL/min,色谱柱半径r单位为cm,色谱柱柱长L单位为cm时,则1个BV/h相当于60QD/(πr
切换时间TS:8~20min;
模拟移动床操作温度:室温;
2)SMBC分离过程如下:
在上述SMBC工作条件下进行SMBC分离,由原料液入口连续泵入喜树果粗提物进样液,SMBC自动控制系统按切换时间TS沿流动相方向将洗脱液入口、萃取液出口、原料液入口和萃余液出口的位置同时依次移动至下一根色谱柱,从萃余液R出口除去各种前杂质,从萃取液E出口得到喜果苷萃取液;
(3)对萃取液进行后处理
将含喜果苷萃取液浓缩至浑浊,静置分层,弃去上层乳白色液体,用蒸馏水对下层沉淀进行清洗,直至清洗液无色;再用乙醇对下层沉淀重结晶,得到喜果苷产品。
上述的一种提纯喜果苷的方法,可以用甲醇替代乙醇作为溶剂或流动相。
喜果苷的检测方法方法为:
高效液相色谱仪:Agilent 1200;分析色谱柱:Extend-C18,5μm,150mm×4.6mmI.D.;流动相:V(甲醇)∶V(水)=60∶40;检测波长:256nm流速:1.2mL/min;柱温:25℃。由喜果苷标准品来标定待测液喜果苷的含量。
下面结合附图用实施例详细描述本发明。
实施例1
1、制备喜树果粗提物
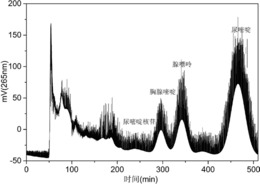
取100g粉碎的喜树果原料,加入1L乙醇,在60℃下超声浸取2次,每次浸取60min,得到浸取液;然后将浸取液浓缩至75mL,加25mLH2O,静置分层,除去该混合液产生的沉淀,取滤液;用水-乙醇(体积比5∶5)稀释滤液,得喜树果粗提物的进样液,其中,喜果苷浓度为0.56mg/mL,其色谱纯度为23.79%,见图1。
2、用SMBC从喜树果粗提物中提纯喜果苷
1)SMBC分离条件如下:
色谱柱:6根,直径1cm,长20cm;
固定相:十八烷基硅烷键合硅胶ODS,20~30μm;
运行模式:1-3-2,即洗脱带:1根色谱柱;精制带:3根色谱柱;吸附带:2根色谱柱,如图2所示;
流动相组成:洗脱带的流动相P为95%乙醇;精制带和吸附带的流动相D为乙醇与水的混合溶液,乙醇的体积百分含量为60%;
流动相流速:在洗脱带中,洗脱液P流速Qp为1.5mL/min,萃取液E流速QE=Qp;在精制带和吸附带中,洗脱液D流速QD为1.53mL/min,进样液F流速QF为0.29mL/min,萃余液R流速QR=QD+QF;
切换时间:9.3min;
模拟移动床操作温度:室温;
2)SMBC分离过程如下:
在上述SMBC工作条件下进行SMBC分离,连续由原料液入口泵入喜树果粗提物的进样液,SMBC自动控制系统按切换时间TS沿流动相方向将洗脱液入口、萃取液出口、原料液入口和萃余液出口的位置同时依次移动至下一根色谱柱,从萃余液R出口除去各种前杂质,萃余液HPLC谱图如图3所示,从萃取液E出口得到喜果苷萃取液,萃取液中喜果苷的色谱纯度为91.56%,收率99.28%,萃取液HPLC谱图如图4所示。
3、对萃取液进行后处理
将含喜果苷萃取液浓缩至浑浊,静置分层,弃去上层乳白色液体,用蒸馏水对下层沉淀进行清洗,直至溶液无色,用乙醇对沉淀三次重结晶,得到色谱纯度为99.1%的喜果苷产品,其HPLC谱图如图5所示,其形貌的扫描电镜照片见图6。
实施例2
除下述内容外,其它条件与步骤均同实施例1;
配制喜树果粗提物进样液:将100g喜树果粉碎后,用乙醇与水(体积比7∶3)的混合溶液60℃下超声浸取二次,每次超声浸取60min,料液比1∶15,得到浸取液;将浸取液浓干后加乙醇与水(体积比7∶3)100mL,得到混合液,除去混合液的沉淀,取滤液;用乙醇与水(体积比5∶5)稀释滤液,得喜树果粗提物的进样液,其中,喜果苷的浓度为1.14mg/mL。
从SMBC萃取液E出口得到喜果苷萃取液,喜果苷的色谱纯度和收率分别为91.07%和99.34%。
对萃取液进行后处理得到色谱纯度为98.80%的喜果苷产品。
实施例3
除下述内容外,其它条件与步骤均同实施例1;
配制喜树果粗提物进样液:将100g喜树果粉碎后,用乙醇与水(体积比7∶3)的混合溶液超声浸取二次,每次超声浸取60min,料液比1∶15,得到浸取液;浸取液浓干后加乙醇与水(体积比6∶4)150mL,得到混合液,除去混合液的沉淀,得滤液;用乙醇与水(体积比6∶4)稀释滤液,得喜树果粗提物的进样液,其中,喜果苷的浓度为1.0mg/mL。
色谱柱:4根,直径2cm,长20cm;
固定相:十八烷基硅烷键合硅胶ODS,30~40μm;
运行模式:1-1-2;
洗脱液P流速Qp为6mL/min,萃取液E流速QE=Qp;洗脱液D流速QD为6mL/min,进样液F流速QF为1.2mL/min,萃余液R流速QR=QD+QF;
从SMBC萃取液E出口,得到喜果苷萃取液,喜果苷色谱纯度91.90%,回收率99.50%。
对萃取液进行后处理得到色谱纯度为98.85%的喜果苷产品。
一种提纯喜果苷的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0