
对苯二酸多晶型和由其获得的喹吖啶酮类化合物")
IPC分类号 : C07C229/62,C07D471/04,C09B48/00,C09B67/00,C09D5/03,C09D11/00






专利摘要
本发明公开了通过在丁二酰丁二酸二甲酯与对甲氧基苯胺的缩合的氧化产物回收期间控制pH而制备2,5-二(对甲氧基苯胺基)对苯二酸结晶I型和II型。所得的2,5-二(对甲氧基苯胺基)-对苯二酸可被转化成具有受控特征的2,9-二甲氧基喹吖啶酮或其固体溶液。
权利要求
1.2,5-二(对甲氧基苯胺基)对苯二酸结晶II型。
2.制造2,5-二(对甲氧基苯胺基)对苯二酸结晶II型的方法,该方法包括将丁二酰丁二酸二甲酯与对甲氧基苯胺缩合,并调整包含缩合产物的反应混合物的pH以实质上立即调整pH到低于约5。
3.制造2,5-二(对甲氧基苯胺基)对苯二酸结晶II型的方法,该方法包括提供具有碱性pH的2,5-二(对甲氧基苯胺基)对苯二酸结晶I型的溶液并调整所述溶液的pH到低于约5.5。
4.制造2,9-二甲氧基喹吖啶酮或其固体溶液的方法,该方法包括将2,5-二(对甲氧基苯胺基)对苯二酸转化成所述2,9-二甲氧基喹吖啶酮或其固体溶液,其中2,5-二(对基苯胺基)对苯二酸是2,5-二(对甲氧基苯胺基)对苯二酸结晶II型。
5.通过权利要求4的方法制造的2,9-二甲氧基喹吖啶酮。
6.通过权利要求4的方法制造的2,9-二甲氧基喹吖啶酮固体溶液。
7.油墨,其包含着色剂和用于着色剂的油墨载体,其中着色剂是权利要求4的方法的产物。
8.权利要求7的油墨,其中着色剂是2,9-二甲氧基喹吖啶酮。
9.权利要求7的油墨,其中着色剂是2,9-二甲氧基喹吖啶酮的固体溶液。
10.油墨,其包含着色剂和用于着色剂的油墨载体,其中着色剂是权利要求3的方法的产物。
11.权利要求10的油墨,其中着色剂是2,9-二甲氧基喹吖啶酮。
12.权利要求10的油墨,其中着色剂是2,9-二甲氧基喹吖啶酮的固体溶液。
13.涂料组合物,其包含着色剂和用于着色剂的涂料组合物载体,其中着色剂是权利要求3的方法的产物。
14.权利要求13的涂料组合物,其中着色剂是2,9-二甲氧基喹吖啶酮。
15.权利要求13的涂料组合物,其中着色剂是2,9-二甲氧基喹吖啶酮的固体溶液。
16.涂料组合物,其包含着色剂和用于着色剂的涂料组合物载体,其中着色剂是权利要求4的方法的产物。
17.权利要求16的涂料组合物,其中着色剂是2,9-二甲氧基喹吖啶酮。
18.权利要求16的涂料组合物,其中着色剂是2,9-二甲氧基喹吖啶酮的固体溶液。
19.着色制品,其包含含有着色剂的基质,其中着色剂是权利要求4的方法的产物。
20.权利要求19的着色制品,其中着色剂是2,9-二甲氧基喹吖啶酮。
21.权利要求19的着色制品,其中着色剂是2,9-二甲氧基喹吖啶酮的固体溶液。
22.着色制品,其包含含有着色剂的基质,其中着色剂是权利要求3的方法的产物。
23.权利要求22的着色制品,其中着色剂是2,9-二甲氧基喹吖啶酮。
24.权利要求22的着色制品,其中着色剂是2,9-二甲氧基喹吖啶酮的固体溶液。
说明书
技术领域技术领域
本发明涉及2,5-二(对甲氧基苯胺基)对苯二酸的新的结晶类型,及其在制备2,9-二甲氧基喹吖啶酮及其固体溶液中的应用。
技术背景背景技术
常规的制备作为2,9-二甲氧基喹吖啶酮或其固体溶液的颜料的方法包括将2,5-二(对甲氧基苯胺基)对苯二酸转化成所需的喹吖啶酮。最终颜料的颜色变换可然后通过改变所选择的合成后的喹吖啶酮生产处理从而改变最终颜料的粒度、粒子形状、粒度分布和/或结晶形式而实现。这些步骤可必然伴有研磨程序(湿法,干法,有和没有研磨助剂)和热处理(有和没有颗粒生长抑制剂或分散助剂)。还已知在合成关环期间使用完全形成的喹吖啶酮衍生物和/或其喹吖啶酮中间物前体来制造喹吖啶酮自身可被用作改变喹吖啶酮的颜色和物理性质的手段。对于本领域的技术人员而言,术语颜料衍生物和衍生物前体是公知的。该衍生物通常被羰基、磺酰基或其它的与含有酸、胺、酰胺、酰亚胺、烷基或者烷氧基的部分反应的官能团取代。
2,5-二(对甲氧基苯胺基)对苯二酸是喹吖啶酮制造中使用的公知的中间体。通常,该中间体如下制备:使丁二酰丁二酸二甲酯和4-甲氧基苯胺在溶剂例如甲醇或高级醇中、在酸存在的条件下、在高温下并可能在压力下反应。得到的二缩合材料与氧化剂诸如间硝基苯磺酸的钠盐、H2O2或空气和碱如NaOH或KOH混合,并随后可能在压力下被加热到高温。得到的2,5-二(4-甲氧基苯胺基)对苯二酸或其金属盐然后用水稀释,获得溶液,可向该溶液中加入过滤助剂如硅藻土并除去不溶物。得到的溶液用酸诸如HCL或H2SO4酸化,直到产物沉淀。
二氢喹吖啶酮(为喹吖啶酮前体)的多晶型,及其对所得喹吖啶酮的影响是公知的。由喹吖啶酮自身表现的多晶型也被辨认。例如,P.V.19多晶型是被最广泛研究和实现商业化的例子之一。其它喹吖啶酮的多晶型诸如例如P.R.122和P.R.202也在文献中被讨论。然而,在文献中没有证明2,5-二苯胺基对苯二酸喹吖啶酮前体表现多晶型。已经假定这些前体仅仅具有一个晶形,并且因为按照推测2,5-二苯胺基对苯二酸溶解在关环剂中,因此它们的物理性质不影响最终获得的喹吖啶酮。
已经发现与其它的2,5-二(苯胺基)对苯二酸衍生物不同,2,5-二(对甲氧基苯胺基)对苯二酸具有两个截然不同的晶形,结晶I型和结晶II型,并且通过选择其中之一,喹吖啶酮或引入2,9-二甲氧基喹吖啶酮的固体溶液的最终性质被改变和/或改善。
发明内容发明概述
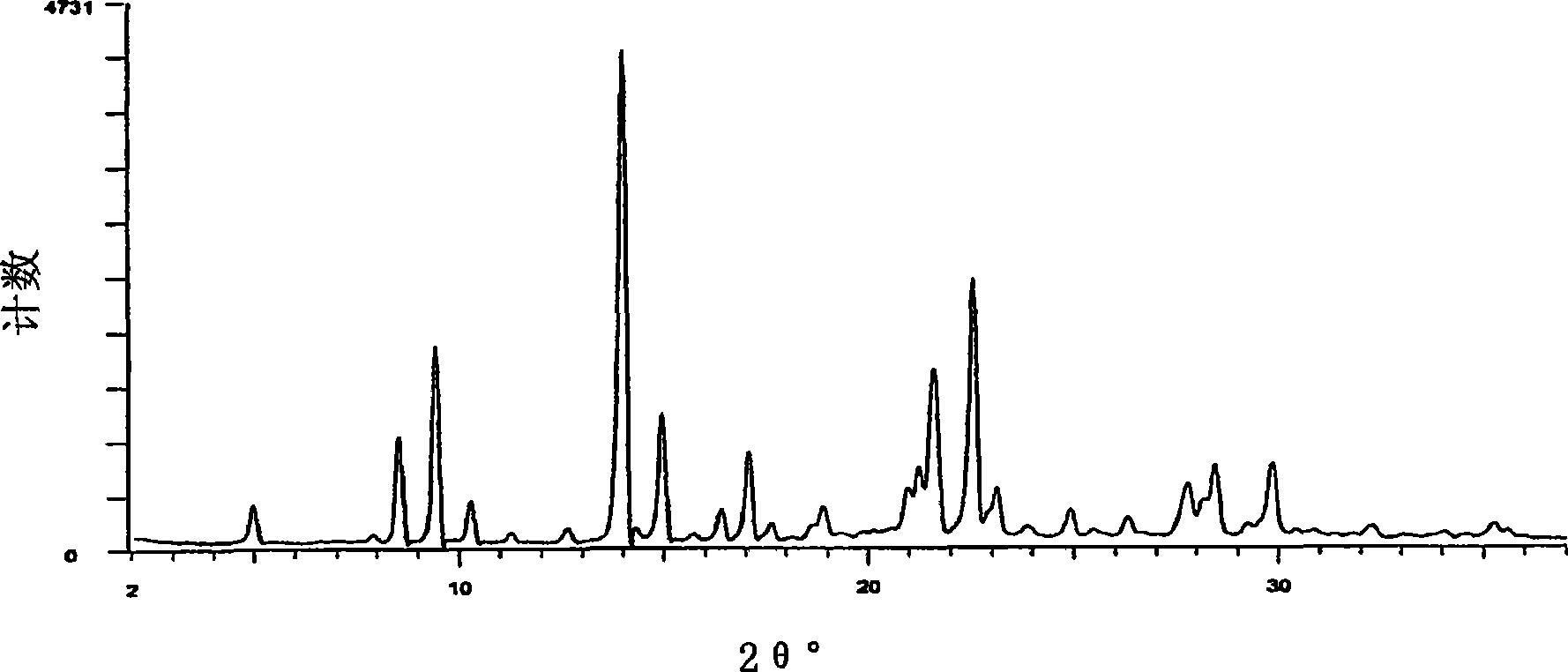
根据本发明,通过控制丁二酰丁二酸二甲酯和4-甲氧基苯胺的被氧化的二缩合产物所经历的pH回收条件,来制造2,5-二(对甲氧基苯胺基)对苯二酸的两个截然不同的晶形,即,结晶I型和结晶II型。结晶I型的特征是褐色(粉末或湿饼)并且具有如图1所示的独特的X射线图形,最强峰在约d=6.3 处(2θ=13.9,CuKa)。通过在产物回收期间第一次调整pH到约中性并然后进一步降低pH到约4.5-约6.5而制造该结晶I型。结晶II型的特征是紫色(粉末或湿饼)并且具有如图2所示的独特的X射线图形,最强峰在约d=16.1 处(2θ=5.5,CuKa)。可以通过向2,5-二(对甲氧基苯胺基)对苯二酸溶液中添加强酸使得最终pH低于约5.0制造结晶II型。作为替代,可将I型转化为II型。
进一步根据本发明,将2,5-二(对甲氧基苯胺基)-对苯二酸转化为2,9-二甲氧基喹吖啶酮或其固体溶液,它们的最终性质相对于现有技术被改变和/或改善。在喹吖啶酮制造期间例如关环期间使用喹吖啶酮中间体的晶形来操控喹吖啶酮的性质。喹吖啶酮在油墨、涂料组合物和大量材料例如塑料中可用作着色剂。
发明详述
本发明提供了2,5-二(对甲氧基苯胺基)对苯二酸的两种截然不同的晶形—结晶I型和结晶II型。这些晶形的差别在于颜色(一种褐色,另一种是紫色)以及X射线衍射图形。还已发现,各自晶形的具有至少约85%的纯度的实质上的纯形式可以如下制备:改变通常使用的沉淀法(即,向2,5-二(对甲氧基苯胺基)对苯二酸的溶液中加入强酸的溶液以实现约<5.0的pH,相对于,使用酸调整2,5-二(对甲氧基苯胺基)对苯二酸的溶液从高pH值到约<7.0的pH)。下文中,褐色晶形是指结晶I型,而紫色晶形是指结晶II型。结晶I型是本领域中先前描述的晶形。
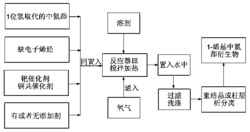
制备本发明的结晶I型或结晶II型的一种方法是在由丁二酰丁二酸二甲酯和对氨基苯甲醚的缩合的被氧化产物的回收期间控制pH。在制备氧化产物的过程中,可将丁二酰丁二酸二甲酯搅拌到液体例如醇中,形成浆料,然后在搅拌下与对氨基苯甲醚混合。加入酸催化剂,将混合物加热至足够的温度和时间以实现缩合。冷却到室温后,可加入氧化剂(例如但不限于间硝基苯磺酸的钠盐,过氧化氢和/或空气)。然后加入碱,将混合物再次加热至足够的温度和时间以实现氧化。当冷却到低于溶剂的回流温度的温度时,将碱性反应浆料用水稀释,并加热到例如约60℃以促进氧化产物在水中的溶解。
为了获得结晶I型(褐色),首先将溶液或浆料的pH从其碱性pH调整到约中性pH,然后进一步调整到约4.5到约6.5。为了获得结晶I1型(紫色),将溶液或浆料与强酸混合,使得所得pH低于约5.0。尽管不束缚于任何理论,据信进行pH调整的时间影响最终的结晶型。结晶II型还可如下制备:提供具有碱性pH的2,5-二(对甲氧基苯胺基)对苯二酸结晶I型的溶液并将该溶液与一定量的足够使pH低于约5.0的酸混合。在所有情况下,所得浆料经过滤和用去离子水洗涤至无传导性状态。
结晶I型的特征在于是褐色的粉末或湿饼,结晶I型的独特的X射线图形如图1所示。结晶II型的特征在于是紫色的粉末或湿饼,并且其独特的X射线图形如图2所示。
过去,忽略了喹吖啶酮颜料前体的晶体形态学是可能影响最终颜料的色彩特性的方式。然而,在本发明中,允许在合成(例如关环)期间使用喹吖啶酮中间体的晶形作为操控所需喹吖啶酮的颜色性质的方法。因此,已经发现在合成2,9-二甲氧基喹吖啶酮及其固体溶液中使用的2,5-二(对甲氧基苯胺基)对苯二酸中间体的晶形诸如但不限于在US专利5,236,498中公开的那些对所得喹吖啶酮的颜色性质有影响,如下表1和2所示。
可通过2,5-二(对甲氧基苯胺基)对苯二酸以及其它的喹吖啶酮中间体例如但不限于2,5-二(苯胺基)对苯二酸、2,5-二(甲苯胺基)对苯二酸和2,5-二(氯苯胺基)-对苯二酸的关环制备固体溶液。也可通过本领域已知的方法诸如但不限于以下的方法制备固体溶液:(1)将粗制颜料组分溶解在强无机酸如硫酸中,然后在其实质上不溶的液体中沉淀,(2)在氧化之前和之后,在强酸或高沸点溶剂中进行喹吖啶酮的合成中间体的关环,然后在其实质上不溶的液体中沉淀,和/或(3)将粗制喹吖啶酮组分一起研磨。
结晶I型和II型中间体可用在任何常规的喹吖啶酮制造过程中,诸如由W.Herbst和K.Hunger的Industrial Organic Pigments(工业有机颜料),由VCH在2004年出版;第452-472页中描述的那些过程中,该文献作为参考并入本文。尽管I型和I1型晶形二者都导致喹吖啶酮在颜色性质方面非常吸引人,但是不同的结晶型可用于影响所得的喹吖啶酮,所得的喹吖啶酮可以是单独的2,9-二甲氧基喹吖啶酮或者是其作为一部分的任何固体溶液,例如但不限于在美国专利5,236,498中公开的固体溶液(作为参考并入本文)。还已经令人惊讶地发现2,5-二(对甲氧基苯胺基)对苯二酸的结晶型可以通过改变色调(shade hue)和色度(chromas)而在颜色方面影响所得的喹吖啶酮。中间体的结晶型还可影响所得的2,9-二甲氧基喹吖啶酮或其固体溶液在以下方面的物理性质:改善表面面积,粒子大小,粒径分布,耐候性,耐晒性和多色调。
当然,中间体的晶形不是影响喹吖啶酮或固体溶液的颜色性质的唯一事件。选择的关环条件也影响对喹吖啶酮颜色所观察的效果和上述讨论到的物理性质,因为它们涉及中间体的晶形。事实上,如果需要,可以选择关环条件从而使得在2,5-二(对甲氧基苯胺基)对苯二酸的褐色和紫色晶形之间观察到的趋势是相反的和/或抵消的。
所得的本发明的颜料或固体溶液颜料可以经历本领域技术人员已知的任何方法的后处理以进一步操控和/或改善颜色、物理性质和耐晒性质。
本发明为颜料制造商提供了新的和有价值的操控最终颜料性质的手段。另外,对这两种晶形及其对最终颜料的颜色性质的影响的认识允许颜料制造商更好地控制工艺过程。为了控制后面步骤的结果,总是有利地制造已知实体。本发明优于现有技术的一个优点是能够制造实质上纯形式的每个晶形,使得制造商更好地控制关环结果。以前,当2,5-二(对甲氧基苯胺基)对苯二酸关环时,没有注意到晶形的纯度,因为人们相信2,5-二苯胺基对苯二酸溶解在多磷酸(或其它适当的关环剂或脱水剂)中,该中间体自身的物理性质还被认为对最终产品的相关性质没有影响。然而,如上所述,已经发现,事实上,2,5-二(对甲氧基苯胺基)对苯二酸实际上是多晶型,而其它通常使用的喹吖啶酮中间体例如2,5-二(苯胺)-对苯二酸(图8)、2,5-二(甲苯胺基)-对苯二酸(图7)和2,5-二(氯苯胺基)-对苯二酸(图9)不是多晶型。另外,已经发现2,5-二(对甲氧基苯胺基)对苯二酸的多晶型影响喹吖啶酮和固体溶液(在其中2,5-二(对甲氧基苯胺基)对苯二酸用作前体或与其它喹吖啶酮前体相组合)的颜色性质。
附图说明附图说明
图1是2,5-二(对甲氧基苯胺基)对苯二酸的结晶I型的X射线图形。
图2是2,5-二(对甲氧基苯胺基)对苯二酸的结晶II型的X射线图形。
图3A-3C是2,5-二(对甲氧基苯胺基)对苯二酸的结晶I型的质谱。
图4A-4C是2,5-二(对甲氧基苯胺基)对苯二酸的结晶II型的质谱。
图5A-5C是已从结晶I型转化来的2,5-二(对甲氧基苯胺基)对苯二酸的结晶II型的质谱。
图6是已从结晶I型转化来的2,5-二(对甲氧基苯胺基)对苯二酸的结晶II型的X射线谱。
图7是通过两种用于获得2,5-二(对甲氧基苯胺基)对苯二酸结晶I型和II型的方法制备的2,5-二(对甲苯胺基)对苯二酸的X射线谱。
图8是通过两种用于获得2,5-二(对甲氧基苯胺基)对苯二酸结晶I型和II型的方法制备的2,5-二(苯胺基)对苯二酸的X射线谱。
图9是通过两种用于获得2,5-二(对甲氧基苯胺基)对苯二酸的结晶I型和II型的方法制备的2,5-二(4-氯苯胺基)对苯二酸的X射线谱。
具体实施方式为了进一步说明本发明,以下描述了多个非限制性实施例,其中(贯穿本说明书和权利要求书),除非另有陈述,否则所有份数和百分数是重量百分数,温度是℃。
实施例1(结晶I型)
向压力反应器中加入489.8克的甲醇,在搅拌下,加入50克的丁二酰丁二酸二甲酯,并搅拌约10分钟。向搅拌的浆料中加入56.7克的对氨基苯甲醚。在搅拌约15分钟后,滴加0.8克的96%H2SO4。密封反应器,加热到95-100℃并在该温度下保持约5小时。冷却到40-50℃后,打开反应器并搅拌,加入55.6克的间硝基苯磺酸的钠盐。搅拌约5分钟后,在15分钟内加入127.8克的45%KOH。密封反应器,加热到90-95℃并在该温度下保持约4小时。冷却到40-50℃后,将反应浆料转移到含800克H2O的容器中。当完成转移后,加入另外275克的H2O并搅拌,将溶液加热到30-40℃。然后使用40.0克的96%H2SO4将pH从13调整到7.5-7.0。在搅拌5分钟后,使用12.8的96%H2SO4将pH进一步调整到5.0-5.5,在30-40℃搅拌15分钟后,过滤所得的褐色浆料并用去离子水洗涤,直到无传导性。干燥该压缩滤饼,获得约85克的褐色粉末。当通过HPLC(装备有996PDA检测器的Waters系统)评价时其纯度为96.3%的2,5-二(对甲氧基苯胺基)对苯二酸。该样品还通过LC-MS(Agilent HP1100 LC/MS)进行评价以通过质谱确定主要组分是2,5-二(对甲氧基苯胺基)对苯二酸。干燥产品的X射线衍射如图1所示,且质谱如图3A-3C所示。样品的元素分析给出以下的分子组成:C=64.77%;H=5.05%;N=6.91%;O=21.94%。
结晶I型的X射线数据(为了简化起见,省略了非常弱的衍射峰)是:
实施例2(结晶II型)
向压力反应器中加入489.8克的甲醇。在搅拌下,加入50克的丁二酰丁二酸二甲酯并搅拌约10分钟,向搅拌的浆料中加入56.7克的对氨基苯甲醚。搅拌约15分钟后,滴加0.8克的96%H2SO4。密封反应器,加热到95-100℃并在该温度下保持约5小时。冷却到40-50℃后,打开反应器并搅拌,加入间硝基苯磺酸的钠盐。搅拌约5分钟后,在15分钟内加入127.8克的45%KOH。密封反应器,在90-95℃加热并在该温度下保持约4小时,冷却到55-60℃后,将反应浆料转移到含有总体积为1200ml的水的容器中。将溶液在55-65℃保持15分钟,然后在45-60分钟内通过泵被转移到包含1344克的3.6%HCL溶液的容器中。所得的紫色浆料在40-50℃搅拌15分钟,此时其pH为1.2。使用15克的50%NaOH将pH调整到pH2-2.5。另外搅拌20分钟后,过滤浆料,紫色压缩饼用水洗涤直到无传导性。干燥该压缩饼获得79.7克的紫色粉末。当通过HPLC评价时,其纯度是97%的2,5-二(对甲氧基苯胺基)对苯二酸。该紫色产品的X射线衍射图形如图2所示。还通过LC-MS评价了该样品以证实主要化合物实际上是2,5-二(对甲氧基苯胺基)对苯二酸,且图4A-4C显示了质谱。样品的元素分析给出以下的分子组成:C=64.76%;H=5.10%;N=6.73%;O=22.63%。
结晶II型的X射线数据(为了简化起见,省略了非常弱的衍射峰)是:
X射线衍射图形(图2)与通过实施例1制造的2,5-二(对甲氧基苯胺基)对苯二酸的X射线衍射图形(图1)的比较显示这两个图形是不同的。
实施例3
将一部分实施例1的褐色产物(50克)溶解在具有碱性pH的水(955克水和75.2克45%KOH)中。所得溶液在55-65℃搅拌60分钟,然后冷却到35-40℃。然后使用31.7克的96%H2SO4将pH从12.8调整到5.3。所得紫色浆料在35-40℃搅拌60分钟,此时将其过滤,所得压缩饼用水洗涤直到无传导性(<120%的incoming wash water)。然后在烘箱中干燥该经洗涤的压缩饼,获得43克的紫色粉末。当通过HPLC评价时,其纯度是93.3%的2,5-二(对甲氧基苯胺基)对苯二酸。通过LC-MS评价证实了主要组分实际上是2,5-二(对甲氧基苯胺基)对苯二酸。质谱(图5A-5C)和X射线衍射图形(图6)证实获得了该紫色类型的中间体。
实施例4(比较例)
向压力反应器中加入300.4克的甲醇,33.9克的丁二酰丁二酸二甲酯,33.9克的对甲苯胺和0.4克的96%H2SO4。密封反应器,并在约1.5小时内加热到92-96℃,然后在92-96℃保持约5小时,冷却到室温后,打开反应器,加入26.2克的间硝基苯磺酸的钠盐以及60.5克的50NaOH和24克的水,密封反应器,并在1.5小时内加热到90-94℃并在该温度下保持约4小时。冷却到室温后,将反应浆料转移到容器中并用水调整总体积到1升,所得浆料在35-45℃搅拌约30分钟,然后使用约96克的25%H2SO4将pH从12调整到5.2,所得的紫色浆料在35-45℃搅拌1小时,此时,其具有的pH为5.5。该浆料过滤并用去离子水洗涤达到20microSiemens。所得压缩饼经干燥,获得约53克的产物,其通过HPLC评价时具有的纯度为>90%的2,5-二(对甲苯胺基)对苯二酸。
实施例5(比较例)
向压力反应器中加入300.4克的甲醇,33.9克的丁二酰丁二酸二甲酯,33.9克的对甲苯胺和0.4克的96%H2SO4。密封反应器,并在约1.5小时内加热到92-96℃,然后在92-96℃保持约5小时,冷却到室温后,打开反应器,加入26.2的间硝基苯磺酸的钠盐和60.5克的50%NaOH和28克的水,密封反应器,并在1.5小时内加热到90-94℃并在90-94℃保持4小时,冷却到室温后,将反应浆料转移到容器中并用水调整到总体积为1升。溶液在40-50℃和pH=12下保持约30分钟,然后通过泵将溶液转移到剧烈搅拌的包含1008克的5.5%H2SO4的容器中。转移需要约1小时并得到与实施例4类似的紫色浆料,该浆料在40-50℃、pH=1.5下搅拌约1小时。将具有pH1.7的浆料过滤并用去离子水洗涤至18microSiemens,所得压缩饼经干燥,得到约53克的产物,其当通过HPLC评价时具有的纯度为>90%的2,5-二(对甲苯胺基)对苯二酸。将实施例4和实施例5的产品的X射线衍射图形重叠,如图7所示,是相同的,表明它们是相同的晶形,即,不是多晶型。
实施例6(比较例)
向压力反应器中加入226克的甲醇,36.0克的丁二酰丁二酸二甲酯,35.3克的苯胺和13.2克的冰醋酸。密封反应器,并在约1.5小时内加热到104℃,然后在104℃保持约5小时,冷却到室温后,打开反应器,加入27克的间硝基苯磺酸的钠盐,74克的50%NaOH和25克的水,密封反应器,并在1.5小时内将反应加热到113℃并在113℃保持约4小时,冷却到63℃后,将反应浆料转移到容器中并用水调整到总体积为1升。所得溶液被加热到40℃并在40℃保持1小时,然后使用145.5克的25%H2SO4将pH从12.2调整到5.3,所得的紫色浆料在40℃搅拌1小时,过滤前的pH是5.4,将浆料过滤并用去离子水洗涤至14microSiemens,所得压缩饼经干燥,获得约52克的产物。当通过HPLC评价时,其纯度为>90%的2,5-二苯胺基对苯二酸。
实施例7(比较例)
向压力反应器中加入226克的甲醇,36.0克的丁二酰丁二酸二甲酯,35.3克的苯胺和13.2克的冰醋酸。密封反应器,并在约1.5小时内加热到104℃,然后在104℃保持约5小时,冷却到室温后,打开反应器,加入27克的间硝基苯磺酸的钠盐,74克的50%NaOH和23克的水,密封反应器,并在1.5小时内将反应加热到113℃并在113℃保持约4小时,冷却到53℃后,将反应浆料转移到容器中并用水调整到总体积为1升,获得的溶液在50℃,pH=12保持1小时。然后通过泵将溶液转移到剧烈搅拌的含1072克的5%H2SO4的容器中,转移需要1小时,得到与实施例6类似的紫色浆料,该浆料在40℃、pH=2.4下搅拌1小时,过滤前pH为2.4,浆料经过滤并用去离子水洗涤至16microSiemens,所得压缩饼经干燥,获得约51.9克的产物。当通过HPLC评价时,其纯度是>90%的2,5-二苯胺基对苯二酸。将实施例7和实施例6的产物的X射线衍射图形重叠进行比较,显示它们是相同的晶形,如图8所示,即,不是多晶型。当通过LC-MS评价时,纯度是95.8%的2,5-二苯胺基对苯二酸。
实施例8(比较例)
向压力反应器中加入303克的,30.3克的丁二酰丁二酸二甲酯,37.9克的4-氯苯胺和0.6克的96%H2SO4。密封反应器,并在约1.5小时内加热到97℃,然后在97℃保持约5小时,冷却到室温后,打开反应器,加入33.4的间硝基苯磺酸的钠盐和77克的50%NaOH和14克的甲醇,密封反应器,并在1.5小时内将反应加热到93℃并在93℃保持约4小时,冷却到34℃后,将反应浆料转移到容器中并调整总体积到1600ml。所得溶液被加热到40℃,并使用146.4克的20%H2SO4在1小时内将pH从12.4调整到5.3。所得的红色浆料在40℃搅拌1小时,过滤前的pH是5.4,将浆料过滤并用去离子水洗涤到45microSiemens,所得的压缩饼经干燥,获得约52.6克的产物。当通过LC-MS评价时,纯度是95.8%的2,5-二(4-氯苯胺基)对苯二酸。
实施例9(比较例)
向压力反应器中加入301.8克的,30.0克的丁二酰丁二酸二甲酯,37.3克的4-氯苯胺和0.6克的96%H2SO4。密封反应器,并在约1.5小时内加热到97℃,然后在97℃保持约5小时,冷却到室温后,打开反应器,加入33.5的间硝基苯磺酸的钠盐和77克的50%NaOH和14.8克的甲醇,密封反应器,并在1.5小时内将反应加热到93℃并在93℃保持约4小时,冷却到室温后,将反应浆料转移到容器中调整总体积到1600ml,以获得溶液,该溶液在50℃、pH=11.8下保持15分钟。然后通过泵将溶液转移到剧烈搅拌的含806克的5.5%H2SO4的容器中,转移需要1.5小时,得到紫色浆料,该浆料在40℃、pH=2下搅拌1小时,在15分钟内,紫色浆料变成红色,浆料经过滤并用去离子水洗涤至16microSiemens,所得压缩饼经干燥,获得约51.5克的产物,当通过LC-MS评价时,纯度为93.7%的2,5-二苯胺基对苯二酸。将实施例9和实施例8的产物的X射线衍射图形重叠进行比较(参见图9),显示具有相同的晶形,即,不是多晶型。
实施例10(得自结晶I型的喹吖啶酮颜料固体溶液)
向容器中加入360.1克的117%多磷酸,然后将其在搅拌下加热到85℃,在85-95℃,在30-60分钟内加入15.2克的,5-二(4-氯苯胺基)对苯二酸,在2,5-二(4-氯苯胺基)对苯二酸已经溶解后,在约90分钟内加入根据实施例1制造的45g的2,5-二(4-甲氧基苯胺基)对苯二酸结晶I型并保持温度<115℃。反应混合物在110-115℃搅拌5小时,然后将反应混合物冷却到约90℃并在约15分钟内慢慢地倾入到剧烈搅拌的含900.6克的甲醇的容器中,在倾入期间通过容器下方的冰-水浴保持甲醇温度<35℃,紫色的甲醇-颜料浆料在室温搅拌1小时,然后被加热至回流(68-72℃),浆料在回流下保持1小时。将浆料冷却到<65℃的温度,然后被倾入到含1350克水的容器中,所得的水/甲醇/颜料的浆料在60℃保持1小时,然后过滤,所得压缩饼用水洗涤到pH3.5。获得含约35.4%固体的135克的压缩饼,得到约48克的固体,从该压缩饼,将67.8克的压缩饼(约24克的干燥颜料)在一定量的足够实现总共170克的水和189克甲醇的水和甲醇中再次成淤浆。用稀NaOH调整浆料的pH到约7.3,然后将其转移到Parr压力反应器中并加入1.5克的50%NaOH,密封反应器并在120-125℃保持6小时,将反应混合物冷却到室温后,将浆料转移到烧杯中,在40-45℃和pH11下,向以上的搅拌浆料中加入溶解在水中的2.0的Dresinate X(由Hercules制造)。在40-45℃持续30分钟后,向浆料中加入溶解在水中的4克的无水氯化钙,在用水调整总体积到1升并在40-45℃搅拌约15分钟后,使用75%的磷酸调整pH到约4.3,在40-45℃搅拌1小时后,将浆料过滤并将所得压缩饼用水洗涤到无传导性的状态,该压缩饼经干燥,获得约25.2克的紫色颜料粉末,该晶形与US专利5,236,498中报道的25%的二氯喹吖啶酮/75%的二甲氧基喹吖啶酮的晶形相同。该颜料的颜色性质当在溶剂型(solventborne)漆料中评价时,如表1和表2所示。
实施例11(得自结晶II型的喹吖啶酮颜料固体溶液)
将360克的117%多磷酸加入到容器中并在搅拌下加热到85℃,在85-95℃,在30-60分钟内加入15.1克的2,5-二(4-氯苯胺基)对苯二酸,在2,5-二(4-氯苯胺基)对苯二酸已经溶解后,在约90分钟内加入根据实施例2制造的45g的2,5-二(4-甲氧基苯胺基)对苯二酸结晶II型,并保持温度<115℃,反应混合物在110-115℃搅拌5小时,然后冷却到约90℃,并在约15分钟内慢慢地被倾入到包含900克的剧烈搅拌的甲醇的容器中。在倾入期间通过容器下方的冰-水浴保持甲醇温度<35℃,紫色的甲醇-颜料浆料在室温下搅拌1小时,然后被加热至回流(68-72℃),浆料在该温度保持1小时,将浆料冷却到<65℃的温度然后倾入到含1350克水的容器中,所得的水/甲醇/颜料浆料在60℃保持1小时,然后过滤,所得压缩饼用水洗涤到pH3.2。获得含约19.94%固体的约274.4克的压缩饼,得到约54.7克的固体,从该压缩饼,将126克的压缩饼(约为25g干重的颜料)在一定量的足够实现总共150克水和150克甲醇的水和甲醇中再次成淤浆,用稀NaOH调整浆料的pH到约7.4,然后将其转移到600mL的Parr压力反应器中并加入1.5克的50%NaOH,密封反应器,并在120-125℃保持6小时,将反应冷却到室温后,将浆料转移到1500毫升的烧杯中,在40-45℃和pH=11.1下,向搅拌的浆料中加入溶解在水中的2.0克的Dresinate X,在30分钟后,加入溶解在水中的4克的无水氯化钙。在用水调整总体积到1升并在40-45℃搅拌约15分钟后,使用75%的磷酸调整pH到约4.3,在40-45℃搅拌1小时后,将浆料过滤并将所得压缩饼用水洗涤到无传导性状态,该压缩饼经干燥,获得约24克的紫色颜料粉末,该晶形与US专利5,236,498中报道的25%的二氯喹吖啶酮/75%的二甲氧基喹吖啶酮的晶形相同。该颜料的颜色性质当在溶剂型漆料中评价时,如表1和表2所示。
实施例12:涂料估价
在相同的溶剂型漆料中合并在相同浓度的实施例10和11的颜料以及根据US 5,236,498制造的颜料(作为标准),然后评价浅色色调和金属色调。与标准相比的结果如表1和2所示。数据显示实施例10的颜料的色调比实施例11中的颜料的色调明显更蓝。
表1-浅色色调
表2-金属色调
实施例13(得自结晶I型的喹吖啶酮颜料)
向容器中加入360克的117的多磷酸并在搅拌下将其加热到85℃,在85-105℃,在60-90分钟内加热根据实施例1制造60克的2,5-二(4-甲氧基苯胺基)对苯二酸结晶I型,反应混合物在110-115℃搅拌5小时,然后将反应混合物冷却到约90-95℃,并在约15分钟内慢慢地倾入到包含900克的剧烈搅拌的甲醇的容器中。在倾入期间通过在圆底烧瓶下方的冰-水浴维持甲醇温度<35℃,将紫色的甲醇-颜料浆料在室温下搅拌1小时,然后加热至回流(68-72℃),将其在该温度保持1小时,将浆料冷却到<65℃,然后倾入到含1350克水的容器中。所得的水/甲醇/颜料浆料在60℃保持3小时,然后过滤,所得压缩饼用水洗涤到pH2.9.,获得含约26.42%固体的约214.3克的压缩饼,得到约56.6克的固体,从该压缩饼,将约25克的压缩饼(约为25克的干重颜料)在一定量的足够实现总共150克水和150克甲醇的水和甲醇中再次成淤浆,用稀NaO小时调整浆料的p小时到约7,然后将其转移到Parr压力反应器中并加入1.5克的50%NaOH,密封反应器,并在120-125℃保持6小时,将反应混合物冷却到室温后,将浆料转移到烧杯中,在40-45℃下,向搅拌的浆料中加入溶解在水中的2.0克的松香皂X。在40-45℃30分钟后,向浆料中加入溶解在水中的4的无水氯化钙,在用水调整总体积为1升后,在40-45℃搅拌约15分钟,用75%的磷酸调整pH到4-4.5,在40-45℃搅拌1小时后,过滤浆料,并用水洗涤所得的压缩饼到无传导性,该压缩饼经干燥,获得约24克的紫色颜料粉末。
实施例14(得自结晶II型的喹吖啶酮颜料)
向容器中加入360克的117%的多磷酸并在搅拌下将其加热到85℃,在85-107℃,在60-90分钟内加入根据实施例2制造的60克的2,5-二(4-甲氧基苯胺基)对苯二酸结晶II型,反应混合物在110-115℃搅拌5小时,然后将反应混合物冷却到约90-95℃,并在约15分钟内慢慢地倾入到包含900克的剧烈搅拌的甲醇的容器中。在倾入期间通过容器下方的冰-水浴维持甲醇温度<35℃,将紫色的甲醇-颜料浆料在室温下搅拌1小时,然后加热至回流(68-72℃),并将其在该温度保持1小时,然后冷却到<65℃的温度,然后倾入到含1350克水的容器中,所得的水/甲醇/颜料浆料在60℃保持3小时,然后过滤,所得压缩饼用水洗涤到p,获得含约45.75%固体的约128.3克的压缩饼,得到约58.7克的固体。从压缩饼将约25克折干计算的颜料再次在足够提供总共150克水和150克甲醇的水和甲醇中成淤浆,用稀NaOH调整浆料的pH到约7,然后将浆料转移到Parr压力反应器中并加入1.5克的50%NaOH,密封反应器,并在120-125℃保持6小时,将反应冷却到室温后,将浆料转移到另一个容器中,在40-45℃下,向搅拌的浆料中加入溶解在水中的2.0克的Dresinate X。在40-45℃30分钟后,向浆料中加入溶解在水中的4克的氯化钙,在用水调整总体积为1升后,在40-45℃搅拌约15分钟,用75%的磷酸调整pH到4-4.5,在40-45℃搅拌1小时后,过滤浆料,并用水洗涤所得的压缩饼到无传导性,该压缩饼经干燥,获得约24克的紫色颜料粉末。
可以对上面描述的优选方案进行各种改变和修改而不脱离本发明的精神和范围。所述实施方案是示例性的而并不对本发明构成任何限制。
2,5-二(甲氧基苯胺基)对苯二酸多晶型和由其获得的喹吖啶酮类化合物专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。










![一种吡咯并[2,1-a]异喹啉衍生物的制备方法](https://www.zhichawang.com/images/CN110759909A/CN110759909A.jpg)


动态评分
0.0