

IPC分类号 : C07D303/26I,C07D407/04I,C07C49/513I,C07C33/14I,C07C35/27I,C07D311/02I,C07D335/06I,C12P17/06I,C12P17/00I,C12P17/02I,C12P7/26I,C12P7/02I,C12P17/16I,A61P19/08I,A61K31/352I,A61K31/382I,A61K31/351I,A61K31/336I,A61K31/122I,A61K31/045I,A







专利摘要
本发明公开了一种萜类衍生物、其顺反异构体、溶剂化物或其药用盐,所述萜类衍生物提取自中国南海东沙软珊瑚Sinulariasandensis的共附生真菌Pseudallescheriaboydii的发酵液。所述萜类衍生物具有抑制或促进破骨细胞生成的活性,并且细胞毒性低,可用于制备抑制或促进破骨细胞生成的药物。本发明为研制新的抑制或促进破骨细胞生成药物提供了先导化合物,为深入研究和开发新的调节破骨细胞生成药物开辟了新的途径,有利于开发利用海洋药用资源。
权利要求
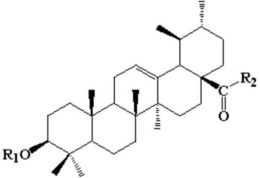
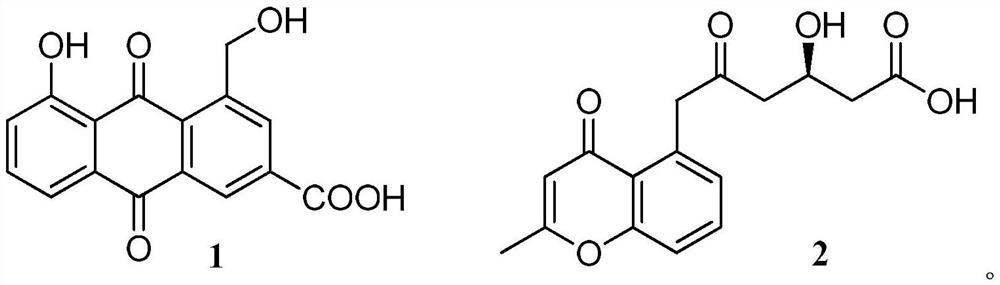
1.一种萜类衍生物或其药用盐,其特征在于:结构如下所示:
2.根据权利要求1所述的萜类衍生物或其药用盐,其特征在于:所述萜类衍生物提取自中国南海东沙软珊瑚Sinularia sandensis的共附生真菌Pseudallescheria boydii的发酵液。
3.一种权利要求1或2所述的萜类衍生物或其药用盐的制备方法,其特征在于:包括以下步骤:
菌株发酵液的制备:将珊瑚Sinularia sandensis的共附生真菌Pseudallescheriaboydii接种至大米培养基,在28℃发酵28天;
将上述获得的大米培养基发酵物溶于乙酸乙酯中按常规进行超声提取,将提取液减压浓缩,得1号粗提物;
将上述1号粗提物分离纯化获得所述萜类衍生物;
所述1号粗提物分离纯化包括以下步骤:
将1号粗提物用正相硅胶色谱分离,依次用石油醚/丙酮=15:1、10:1、5:1、3:1,二氯甲烷/甲醇=30:1、20:1、15:1、10:1、8:1、5:1、100%丙酮洗脱液进行梯度洗脱,根据薄层板监测,按照各流份极性的差别从小到大共收集13个部分洗脱液,以馏分A-馏分M即Fr.A-Fr.M表示;
将Fr.F经过Sephadex LH-20凝胶柱层析得到5个组分Fr.F.1~Fr.F.5,将Fr.F.4经正相硅胶柱层析及半制备高效液相得到化合物8和化合物9;
将Fr.G经过Sephadex LH-20凝胶柱层析得到6个组分Fr.G.1~Fr.G.6,将Fr.G.6经正相硅胶柱层析及半制备高效液相得到化合物6;
将Fr.L经过Sephadex LH-20凝胶柱层析得到4个组分Fr.L.1~Fr.L.4,将Fr.L.3经正相硅胶柱层析及半制备高效液相得到化合物10;
所述大米培养基的配方:1000g大米,800mL人造海水,200mL蒸馏水。
4.一种萜类衍生物或其药用盐在制备抑制或促进破骨细胞生成的药物中的应用,其特征在于,所述萜类衍生物的结构如下所示:
5.一种药物组合物在制备抑制或促进破骨细胞生成药物中的应用,其特征在于,所述药物组合物包括权利要求4所述的萜类衍生物或其药用盐以及药学上可接受的载体。
说明书
技术领域
本发明属于医药技术领域,尤其涉及一种萜类衍生物及其制备方法与应用。
背景技术
宿主珊瑚Sinularia sandensis属于软珊瑚目(Alcyonacea)、Alcyoniina亚目、Alcyoniidae科、Sinularia属动物。目前对于软珊瑚Sinularia sandensis的研究较少,仅有3篇文献报导,对其共附生微生物化学成分的研究有1篇文献报导。目前,从该种珊瑚中分离得到的化合物,主要为西松烷型二萜和倍半萜类化合物(Anjaneyulu A;RaoGV;Sagar K;KumarKR;MohanKC,Sandensolide:A new dihydroxycembranolide from the soft coral,Sinularia sandensis Verseveldt of the Indian Ocean.Natural Product Letters1995,7(3),183-190;Tsai TC;ChenHY;Sheu JH;ChiangMY;WenZH;DaiCF;SuJH,StructuralElucidation and Structure–Anti-inflammatory Activity Relationships ofCembranoids from Cultured Soft Corals Sinularia sandensis and Sinulariaflexibilis.Journal of agricultural and food chemistry 2015,63(32),7211-7218;ChengSY;WangSK;ChenPW;DuhCY,Sandensone A,a novel sesquiterpenoid from theFormosan soft coral Sinularia sandensis.Bioorganic&medicinal chemistryletters 2015,25(11),2353-2355)。从该种珊瑚共附生微生物中分离得到的化合物,主要为十氢化萘类化合物(SunYZ;Tibor K;Attila M;Tang H;Chou Y;Soong K;Su L;Sun P;Zhuang C L;Zhang W,Immunomodulatory Polyketides from a Phoma-like FungusIsolated from a Soft Coral.Journal of Natural Products,2017,80(11):2930-2940)。
共附生真菌Pseudallescheria boydii属于子囊菌门(Ascomycota)、粪壳菌纲(Sordariomycetes)、肉座菌亚纲(Hypocreomycetidae)、小囊菌目(Microascales)、小囊菌科(Microascaceae)、Pseudallescheria属,通常处于死水和污染水体中。目前,从该种真菌中分离得到的化合物种类丰富,包括聚酮、哌嗪、萜类、多肽、生物碱和吡嗪类似物等。(Chang Y C,Deng T S,Pang K L,et al.Polyketides from the littoral plantassociated fungus Pseudallescheria boydii.Journal of Natural Products2013,76(9):1796-1800;Lan W J,Liu W,Liang W L,et al.Pseudaboydins A and B:NovelIsobenzofuranone Derivatives from Marine Fungus Pseudallescheria boydiiAssociated with Starfish Acanthaster planci.Marine Drugs2014,12(7):4188–4199;Lan W J,Wang K T,Xu M Y,et al.Secondary metabolites with chemical diversityfrom the marine-derived fungus Pseudallescheria boydii F19-1and theircytotoxic activity.Rsc Advances2016,6(80):S1-S117;Su H J,Lin M J,YiJung T,etal.Pseudallin,a new antibiotic produced by the human pathogenic fungusPseudallescheria boydii,with ecological significance.Botanical Studies2012,53(2):239-242;Nirma C,Eparvier V,Stien D.Antifungal agents fromPseudallescheria boydii SNB-CN73isolated from a Nasutitermessp.termite.Journal of Natural Products2013,76(5):988-992;Wu Q,Jiang N,Bo H W,et al.Antibacterial epipolythiodioxopiperazine and unprecedentedsesquiterpene from Pseudallescheria boydii,a beetle coleoptera-associatedfungus.Organic&Biomolecular Chemistry2014,12(46):9405-9412;Sorres J,Nirma C,Eparvier V,et al.Pseudallicins A-D:Four Complex Ovalicin Derivatives fromPseudallescheria boydii SNB-CN85.Organic Letters2017,19(15):3978-3981)。但是,未见有关从该种真菌中分离得到所述萜类衍生物以及此类化合物具有调节破骨细胞生成的报道。
发明内容
本发明的第一个目的是提供一种萜类衍生物。
本发明的第二个目的是提供一种所述萜类衍生物的制备方法。
本发明的第三个目的是提供一种所述萜类衍生物在制备调节破骨细胞生成的药物中的应用。
为了实现上述目的,本发明采用的技术方案如下:
本发明的第一个方面提供了一种萜类衍生物、其顺反异构体、溶剂化物或其药用盐,结构如下所示:
所述萜类衍生物提取自中国南海东沙软珊瑚Sinularia sandensis的共附生真菌Pseudallescheria boydii的发酵液。
所述药用盐为有机酸、无机酸盐。
所述无机酸是指盐酸、硫酸、磷酸、二磷酸、氢溴酸或硝酸等。
所述有机酸是指乙酸、苹果酸、马来酸、柠檬酸、富马酸、酒石酸、琥珀酸、乳酸、对甲苯磺酸、水杨酸或草酸等。
所述药用盐还包括药物可接受的载体、赋形剂或者辅料。
本发明的第二个方面提供了一种所述萜类衍生物、其顺反异构体、溶剂化物或其药用盐的制备方法,包括以下步骤:
菌株发酵液的制备:将珊瑚Sinularia sandensis的共附生真菌Pseudallescheria boydii分别接种至大米培养基及生物麦芽提取物琼脂培养基,在28℃发酵28天;
将上述获得的大米培养基发酵物溶于乙酸乙酯中按常规进行超声提取,将提取液减压浓缩,得1号粗提物;
将上述生物麦芽提取物琼脂培养基发酵物溶于乙酸乙酯中按常规进行超声提取,将提取液减压浓缩,得2号粗提物;
将上述1号粗提物和2号粗提物分离纯化获得所述萜类衍生物。
所述1号粗提物分离纯化包括以下步骤:
将1号粗提物用正相硅胶色谱分离,依次用石油醚/丙酮=15:1、10:1、5:1、3:1,二氯甲烷/甲醇=30:1、20:1、15:1、10:1、8:1、5:1、100%丙酮洗脱液进行梯度洗脱,根据薄层板监测,按照各流份极性的差别从小到大共收集13个部分洗脱液,以馏分A-馏分M即Fr.A-Fr.M表示;
将Fr.D经过Sephadex LH-20凝胶柱层析得到4个组分Fr.D.1~Fr.D.4,将Fr.D.4经正相硅胶柱层析及半制备高效液相得到化合物3;
将Fr.F经过Sephadex LH-20凝胶柱层析得到5个组分Fr.F.1~Fr.F.5,将Fr.F.4经正相硅胶柱层析及半制备高效液相得到化合物2、化合物8和化合物9,半制备高效液相得到化合物1;
将Fr.G经过Sephadex LH-20凝胶柱层析得到6个组分Fr.G.1~Fr.G.6,将Fr.G.6经正相硅胶柱层析及半制备高效液相得到化合物6;
将Fr.L经过Sephadex LH-20凝胶柱层析得到4个组分Fr.L.1~Fr.L.4,将Fr.L.3经正相硅胶柱层析及半制备高效液相得到化合物10。
所述2号粗提物分离纯化包括以下步骤:
将2号粗提物用正相硅胶色谱分离,依次用石油醚/丙酮=15:1、10:1、5:1、2:1,二氯甲烷/甲醇=40:1、20:1、15:1、10:1、7:1、3:1、100%丙酮洗脱液进行梯度洗脱,根据薄层板监测,按照各流份极性的差别从小到大共收集14个部分洗脱液,以馏分A-馏分J即Fr.A-Fr.J表示;
将Fr.B经过Sephadex LH-20凝胶柱层析得到6个组分Fr.B.1~Fr.B.6,将Fr.B.4经正相硅胶柱层析及制备高效液相得到化合物1、化合物4、化合物5和化合物7。
所述大米培养基的配方:1000g大米,800mL人造海水,200mL蒸馏水。
所述生物麦芽提取物琼脂培养基的配方:20gBiomalt,15g琼脂,800mL人造海水,200mL蒸馏水。
本发明的第三个方面提供了一种所述萜类衍生物、其顺反异构体、溶剂化物或其药用盐在制备抑制或促进破骨细胞生成的药物中的应用。
本发明的第四个方面提供了一种药物组合物,包括所述萜类衍生物、其顺反异构体、溶剂化物或其药用盐以及药学上可接受的载体。
本发明的第五个方面提供了一种所述药物组合物在制备抑制或促进破骨细胞生成药物中的应用。
由于采用上述技术方案,本发明具有以下优点和有益效果:
本发明从珊瑚Sinularia sandensis的共附生真菌Pseudallescheria boydii大米培养基及生物麦芽提取物琼脂培养基发酵液中提取分离得到的萜类衍生物,通过红外、紫外、质谱和二维核磁共振等多种现代光谱技术的综合解析进行结构鉴定,确定了所述化合物的化学结构。所述萜类衍生物具有抑制或促进破骨细胞生成的活性,并且细胞毒性低,可用于制备抑制或促进破骨细胞生成的药物。本发明为研制新的抑制或促进破骨细胞生成药物提供了先导化合物,为深入研究和开发新的调节破骨细胞生成药物开辟了新的途径,有利于开发利用海洋药用资源。
附图说明
图1为本发明实施例制备的化合物1-10对破骨细胞生成的作用中,测量不同浓度的化合物的破骨细胞的面积和数量的示意图,其中:A为剂量为0.1μM药物的破骨细胞的数量示意图,B为剂量为1μM药物的破骨细胞的数量示意图,C为剂量为0.1μM药物的破骨细胞的面积示意图,D为剂量为1μM药物的破骨细胞的面积示意图。
图2为本发明实施例制备的化合物2、化合物4、化合物7、化合物10,剂量分别为0.1μm和1μm时破骨TRAP染色图。
具体实施方式
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
本发明从珊瑚Sinularia sandensis的共附生真菌Pseudallescheria boydii的大米培养基和生物麦芽提取物琼脂培养基发酵液中提取分离得到10个萜类衍生物,通过红外、紫外、质谱和二维核磁共振等多种现代光谱技术的综合解析进行结构鉴定,确定了这10种化合物的化学结构。
实施例1
实施例所用菌株发酵液的制备:将珊瑚Sinularia sandensis的共附生真菌Pseudallescheria boydii分别接种至大米培养基及生物麦芽提取物琼脂培养基,在28℃发酵28天。
所述大米培养基的配方:1000g大米,800mL人造海水,200mL蒸馏水。
所述生物麦芽提取物琼脂培养基的配方:20gBiomalt,15g琼脂,800mL人造海水,200mL蒸馏水。
制备化合物1-10:
1.制备总粗提物
1.1将上述获得的6KG大米培养基发酵物溶于乙酸乙酯中按常规进行超声提取(发酵物捣碎成小块,15L乙酸乙酯超声提取5次,每次30min),将提取液减压浓缩,得1号粗提物60.4g。
1.2将12L上述生物麦芽提取物琼脂培养基发酵物溶于乙酸乙酯中按常规进行超声提取(发酵物捣碎成小块,12L乙酸乙酯超声提取5次,每次30min),将提取液减压浓缩,得2号粗提物5.6g。
2.分离纯化
2.1将1号粗提物用正相硅胶色谱分离,依次用石油醚/丙酮=15:1、10:1、5:1、3:1,二氯甲烷/甲醇=30:1、20:1、15:1、10:1、8:1、5:1、100%丙酮洗脱液进行梯度洗脱,根据薄层板监测,按照各流份极性的差别从小到大共收集13个部分洗脱液,以馏分A-馏分M(Fr.A-Fr.M)表示;
将Fr.D经过Sephadex LH-20凝胶柱层析(葡聚糖凝胶,美国Pharmacia公司)(二氯甲烷/甲醇=2:1)得到4个组分(Fr.D.1~Fr.D.4),将Fr.D.4经正相硅胶柱层析(石油醚:丙酮=30:1)及半制备高效液相(甲醇/水=65/35,流速为2mL/min)得到化合物3(1.0mg,保留时间70min)。
将Fr.F经过Sephadex LH-20凝胶柱层析(二氯甲烷/甲醇=2:1)得到5个组分(Fr.F.1~Fr.F.5),将Fr.F.4经正相硅胶柱层析(石油醚:丙酮=15:1)及半制备高效液相(甲醇/水=45/55,流速为1.5mL/min)得到化合物2(5.4mg,保留时间22min)、化合物8(5.6mg,保留时间46min)和化合物9(3.5mg,保留时间50min),半制备高效液相(甲醇/水=56/34,流速为2mL/min)得到化合物1(4.2mg,保留时间30min)。
将Fr.G经过Sephadex LH-20凝胶柱层析(二氯甲烷/甲醇=2:1)得到6个组分(Fr.G.1~Fr.G.6),将Fr.G.6经正相硅胶柱层析(二氯甲烷:甲醇=300:1)及半制备高效液相(甲醇/水=56/44,流速为1.5mL/min)得到化合物6(4.7mg,保留时间57min)。
将Fr.L经过Sephadex LH-20凝胶柱层析(二氯甲烷:甲醇=2:1)得到4个组分(Fr.L.1~Fr.L.4),将Fr.L.3经正相硅胶柱层析(二氯甲烷:甲醇=20:1)及半制备高效液相(甲醇/水=42/58,流速为2mL/min)得到化合物10(1.6mg,保留时间52min)。
2.2将2号粗提物用正相硅胶色谱分离,依次用石油醚/丙酮=15:1、10:1、5:1、2:1,二氯甲烷/甲醇=40:1、20:1、15:1、10:1、7:1、3:1、100%丙酮洗脱液进行梯度洗脱,根据薄层板监测,按照各流份极性的差别从小到大共收集14个部分洗脱液,以馏分A-馏分J(Fr.A-Fr.J)表示。
将Fr.B经过Sephadex LH-20凝胶柱层析(二氯甲烷:甲醇=2:1)得到6个组分(Fr.B.1~Fr.B.6),将Fr.B.4经正相硅胶柱层析(石油醚/丙酮=10:1)及制备高效液相(甲醇/水=67/33,流速为18mL/min)得到化合物1(7.4mg,保留时间45min)、化合物4(2.6mg,保留时间64min)、化合物5(3.6mg,保留时间68min)和化合物7(5.6mg,保留时间77min)。
3.结构鉴定
通过红外、紫外、质谱和二维核磁共振等多种现代光谱技术的综合解析进行结构鉴定,确定了化合物1-10的化学结构。
化合物1:制备、表征见文献KubokiH;TsuchidaT;WakazonoK;IsshikiK;KumagaiH;YoshiokaT,J.Antibiot.1999,52,590–593.
化合物2:制备、表征见文献:Takamatsu S;Kim YP;Komiya T;Sunazuka T;Hayashi M;TanakaH;KomiyamaK,J.Antibiot.1996,49,635–637.
化合物3:制备、表征见文献:Zhang P;Bao B;Dang H T;Hong J;Lee H J;Yoo ES;Bae KS;Jung J H,J.Nat.Prod.2009,72,270–275.
化合物4:制备、表征见文献:Yaginuma A;Ishikawa S,Jpn.Patent 09309859,1997;Chem.Abstr.1998,128,74385.
化合物5:制备、表征见文献:DeshengL;YulingH;ChunminL;LiyingM;XiaohongP;DaneelF;WeizhongL,Rec.Nat.Prod.2016,10,708–713.
化合物6:无色油状物;Rf=0.43(二氯甲烷/甲醇=15:1); UV(CH3CN)λmax:195(2.139)nm;ECD(CH3CN,c 0.0031M)λmax(Δε)219(+0.19)nm;IR(film)νmax 3407,2977,2930,1770,1647,1386,1252,1182,1054cm
化合物7:灰白色非结晶固体;Rf=0.41(石油醚/丙酮=3:1) UV(CH3CN)λmax:<190nm;ECD(CH3CN,c 0.00159M)λmax(Δε):293(+1.32),204(-0.91)nm;IR(film)νmax 3481,2966,2923,1726,1665,1377,1108,1063,876cm
化合物8:无色晶体;m.p.219.2-222.0℃;Rf=0.69(二氯甲烷/甲醇=20:1); UV(CH3CN)λmax:194nm;ECD(CH3CN,c 0.00087M)λmax(Δε):291(+1.21,205(+9.51);IR(film)νmax 3429,2964,2923,1731,1639,1382,1097,1061,1006,896cm
化合物9:灰白色非结晶固体;Rf=0.62(二氯甲烷/甲醇=20:1); UV(CH3CN)λmax:197nm;ECD(CH3CN,c 0.00287M)λmax(Δε):216(+0.74),202.5(+1.75)nm;IR(film)νmax 3367,2926,2855,1705,1664,1449,1383,1089,1008,899cm
化合物10:无色油状液体;Rf=0.45(二氯甲烷/甲醇=15:1); UV(CH3CN)λmax:191.80nm;HR-ESI-MS m/z 305.1507[M+Cl]
表1.化合物4,6的核磁共振氢、碳谱数据(in CDCl3)
表2.化合物7-9的核磁共振氢谱数据(in CDCl3)
表3.化合物7-9的核磁共振碳谱数据(in CDCl3)
表4.化合物10的核磁共振碳谱数据(in CDCl3)
实施例2
本发明化合物1~10对破骨细胞生成调节实验
1.实验用动物株
雌性C57BL/6小鼠(18-20g)购自上海斯莱克实验动物有限公司。所有实验操作均严格按照国家对实验动物的使用和保养规定进行,符合作者所在单位的实验动物中心确定的指导方针,并通过第二军医大学动物伦理委员会批准。
2.实验试剂、耗材和仪器
眼科剪、眼科镊三套;高压蒸汽灭菌锅;培养皿3个;5ml无菌注射器一个;T75瓶一个;α-mem;gbico FBS(10%);PS(1%);二甲基亚砜DMSO(sigma);移液管(Corning);96孔板(Corning);CO2孵箱(SANYO);TRAP染色剂。
3.实验用药
本发明化合物1-10由实施例1制备。
4.小鼠骨髓巨噬细胞(BMMs)制备
(1)选取4-6周C57小鼠,雌雄皆可,将C57BL/6小鼠颈部脱臼处死,泡在酒精中10-15分钟,拿出小鼠放在第一个皿中,用第一套剪、镊将双侧下肢皮肤切开,找到髂骨位置,将整个下肢剪下,注意保护股骨头的完整性。
(2)用第二套剪子、镊子将股骨、胫骨分开,周围的肌肉尽量剔除干净,放在第二个皿中。
(3)用第三套剪子、镊子将股骨、胫骨两侧剪开,用注射器吸取提前配好的培养基,插入骨髓中冲洗骨髓腔1-2次,见骨髓腔由红变白后即可。
(4)将冲洗出来的细胞吸取到15ml离心管中,1000r、5min,弃去上清液,加入1-2ml培养基重悬,置入T75瓶中,加入10ml培养基,培养三天。
(5)细胞培养第三天换液,观察细胞状态,第四天胰酶消化5min,用细胞刮将细胞刮下,1000r、5min离心重悬并细胞计数。
(6)96孔板种细胞,密度为8000/well,每孔100ul含巨噬细胞集落刺激因子(M-CSF)的培养基(M-CSF的最终浓度为30ng/ml),种细胞的孔周围加一圈PBS以减少边缘效应。
(7)种板后第二天,细胞贴壁后,吸取原培养基并加入相应培养基及药物(化合物1~10)。药物浓度为1μM、0.5μM;每孔100ul含M-CSF和破骨细胞分化因子(RANKL)的培养基(M-CSF最终浓度为30ng/ml,RANKL最终浓度为100ng/ml),每两天换一次含药物的培养基。
(8)换第三次药物(种板后第5-6天)时观察,如果对照组出现破骨细胞,则终止加药,吸取培养基并加入4%多聚甲醛100ul/well固定10min,吸取固定液,用pbs洗三遍,加入50ul TRAP染色剂/well,培养箱孵育46-60min,看到细胞染成粉红色即可,吸取染色剂,加入pbs100ul/well,显微镜下观察并拍照。
二、实验结果
对比对照组和本发明化合物1-10处理组的抑制破骨细胞生成模型,分析本发明化合物1~10对抑制破骨细胞生成的影响。如图1所示,图1为本发明实施例制备的化合物1-10对破骨细胞生成的作用中,测量不同浓度的化合物的破骨细胞的面积和数量的示意图,其中:A为剂量为0.1μM的药物的破骨细胞的数量示意图,B为剂量为1μM药物的破骨细胞的数量示意图,C为剂量为0.1μM药物的破骨细胞的面积示意图,D为剂量为1μM药物的破骨细胞的面积示意图。图中编号1-10对应化合物1-10,control为空白组,这10种化合物在0.1μM和1μM浓度条件下对破骨细胞的细胞毒性很低。
在上述实验条件的基础上,分别用上述浓度处理骨髓原代细胞,在进行破骨细胞调节实验中,对照组和处理组均加入了促进骨髓原代细胞生长的巨噬细胞集落刺激因子(M-CSF)药物和诱导细胞向破骨细胞生成的破骨细胞分化因子(RANKL)药物,6天后如图2所示,图2为本发明实施例制备的化合物2、化合物4、化合物7、化合物10,剂量分别为0.1μm和1μm时破骨TRAP染色图。与对照组相比,control为空白组,化合物4(compound-4)、化合物7(compound-7)处理组在0.1μM和1μM的药物浓度下破骨细胞出现的数量显著增加,但破骨细胞出现的面积仅在0.1μM药物浓度时才增加,说明化合物4和化合物7明显促进破骨细胞生成;化合物6(compound-6)处理组在1μM药物浓度时破骨细胞出现的数量增加,说明化合物6对促进破骨细胞生成有一定作用;化合物2(compound-2)处理组在0.1μM和1μM的药物浓度下破骨细胞出现的数量减少,但破骨细胞出现的面积仅在0.1μM药物浓度时才减少,说明化合物2具有一定的抑制破骨细胞生成的作用。化合物10(compound-10)处理组只在0.1μM的药物浓度下破骨细胞出现的数量增加,说明化合物10具有一定的促进破骨细胞生成的作用。上述实验结果表明,本发明化合物具有抑制或促进破骨细胞生成的活性,故可用于制备抑制或促进破骨细胞生成的药物。本发明为研制新的抑制或促进破骨细胞生成药物提供了先导化合物,为深入研究和开发新调节破骨细胞生成的药物开辟了新的途径,有利于开发利用海洋药用资源。
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
一种萜类衍生物及其制备方法与应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。













动态评分
0.0