










专利摘要
公开了式(A)和(B)所示的化合物。本发明的化合物可用于改善治疗用阿片剂的副作用。
权利要求
1.一种化合物,所述化合物具有下式:
其中,Y为抗衡离子。
2.根据权利要求1所述的化合物在制备用于改善给药阿片剂的患者的阿片剂副作用的药物中的应用。
3.根据权利要求2所述的应用,其中,所述副作用选自便秘、恶心/呕吐、咳嗽抑制、瘙痒、烦躁不安和尿潴留。
4.根据权利要求1所述的化合物在制备用于改善已经历手术的患者的手术后肠功能的药物中的应用。
说明书
联邦赞助的研究
以下发明是在国家健康学会(NIH)/国家药品滥用研究所(NIDA)授予的合同R01DA12180下,由政府支持完成的发明。政府对此发明享有一定的权力。
联合研究协议
本申请里所阐述的发明是由Mark Wentland(伦勒斯工业学院)及Alkermes有限公司或者以他们的名义来完成的,他们是在发明之日或之前生效的联合研究协议里的协议方,本发明是在联合研究协议的范围内进行工作的成果。
相关申请的交叉引用
本申请要求2007年8月9日提交的美国临时申请60/954,960的优先权,其全部内容在此引入作为参考。
技术领域
本发明涉及可用于改善治疗用阿片剂(opiate)的外周副作用(peripheralside effect)的阿片样物质(opioid)受体结合化合物。
背景技术
自从1805年吗啡的分离以来,阿片剂就已成为频繁研究的主题,已经有数千种具有阿片剂或类阿片剂活性的化合物被鉴定。许多与类阿片样物质受体相互作用的化合物,包括用于产生痛觉缺失的化合物(例如,吗啡)和用于治疗药瘾的化合物(例如,纳曲酮和环佐辛),由于口服生物利用度差和从身体的清除速率非常快,因此对人具有有限的效用。这表明,在许多情况下是由于在2,6-亚甲基-3-苯并吖辛因(2,6-methano-3-benzazocines)(也称为苯并吗吩烷(benzomorphan)[(例如,环佐辛和EKC(乙基酮基环佐辛)])中存在8-羟基(OH)和在吗啡喃(morphinane)(例如,吗啡)中存在相应的3-OH基。
苯并吗吩烷编号 吗啡喃编号
这些羟基的高极性延迟了母体分子的口服吸收。此外,8-(或3-)OH基易于磺化和葡萄糖苷酸化(II期代谢),二者均促进活性化合物的快速排泄,导致活性化合物的半衰期不利地变短。直至2001年Wentland的报导,本领域在过去七十年的一致经验是除去或替换8-(或3-)OH基产生药理学非活性化合物。
美国专利6,784,187(授权给Wentland)公开了阿片样物质的酚OH基可被替换为CONH2。在阿片样物质的环佐辛系列中,已表明8-酰胺基环佐辛(8-CAC)对于μ和κ阿片样物质受体具有高亲和力。在体内研究中,8-CAC显示高抗伤害性疼痛的活性(antinociception activity),且当在小鼠中均以1mg/kg的剂量腹腔给药时,8-CAC作用的持续时间比环佐辛长得多(8-CAC为15小时,而环佐辛为2小时)。
已公开了用于预防或缓解用于抑制麻醉性镇痛药(例如吗啡和相关的阿片剂)的副作用的肠移动、而不会削弱该麻醉性镇痛药的镇痛活性的阿片拮抗剂纳曲酮的季衍生物。例如,在美国专利4,176,186(Goldberg等人)中公开了甲基纳曲酮。但是,预防或抑制用于抑制副作用的肠运动所需的剂量较高。因此,需要开发在较低剂量下具有提高的活性的化合物。
发明内容
本发明的发明人现已发现,通过使8-(或3-)羟基被多种小的、极性、中性残基(如本文所定义,羟基和低级烷氧基除外)例如酰胺基、硫代酰胺基、羟基脒基和甲酰胺基替换,可制备纳曲酮季铵盐(quaternary naltrexone salt)的衍生物。此外,本发明的发明人还发现,酰胺的氮可被相当大且相对非极性的基团取代。所有这些化合物表现出优异的阿片样结合性且具有良好至优异的外周阿片拮抗活性。不仅苯并吗吩烷(benzomorphan)如此,本文所述的吗啡喃酰胺对阿片样物质受体具有高亲和力,本文所述的含有小的、极性、中性残基(例如酰胺基、硫代酰胺基、羟基脒基和甲酰胺基)替换OH基的化合物作用时间长、对II期代谢远不敏感,且通常更容易口服生物利用。
本发明化合物可用于改善治疗用阿片剂的副作用。这些副作用包括便秘、恶心/呕吐(吐出)、咳嗽抑制、瘙痒、烦躁不安和尿潴留。本发明化合物还可用于改善可能与阿片样物质治疗相关或者可能无关的手术后肠功能。
在一个方面,本发明涉及具有下式所示的2,6-亚甲基-3-苯并吖辛因-8-酰胺和2,6-亚甲基-3-苯并吖辛因-8-酰胺衍生物:
其中,
G为选自极性、中性残基,特别是,可选自由以下基团组成的组:取代或未取代的酰胺基(amide group),包括但不限于酰胺基(carboxamide group)、硫代酰胺基、酰基胺基和甲酰胺基;取代或未取代的胺;取代或未取代的脒,例如羟基脒;和被极性中性残基取代的烷基。优选G可选自CONH2和CSNH2。
例如,G可为-CH2Z(其中Z为极性中性残基,例如CH2ORa、CH2NRbRc)、-CN、-NRbSO2-Rc、-C(=W)Ra、-NRaCORb、-NRaCSRb、-SO2NRbRc、-NRb-Qa-Rc、(C=W)NRbRc、C(O)ORa、杂环、取代的杂环、杂芳基和取代的杂芳基,例如 其中,1为0、1、2、3、4或5;k为0、1或2;X为C、N、S或O,且 代表单键或双键;
Ra、Rb、Rc各自独立地选自:
氢;
芳基;取代的芳基;杂芳基;取代的杂芳基;
杂环或取代的杂环;和
各自含有0、1、2或3个或更多个选自O、S或N的杂原子的取代或未取代的烷基、链烯基、炔基、环烷基或环烯基;
或者,Ra、Rb和Rc与相连的原子共同形成杂环或取代的杂环;
Qa不存在或选自(C=O)、(SO2)、(C=NH)、(C=S)或(CONRa);
W为O、S、NORa或NRa;
Y为药学上可接受的抗衡离子;例如,Y可以为酒石酸盐、柠檬酸盐、盐酸盐或甲磺酸盐;
Q为取代或未取代的、饱和或不饱和的脂肪族或芳香族基团,例如取代或未取代的、饱和或不饱和的烷基(例如,C1-C20-烷基)、链烯基(例如,C2-C20-链烯基)、炔基(例如,C2-C20-炔基)芳基、杂芳基、杂环基、芳基烷基(例如,其中芳基为C6-C10-芳基且烷基为C1-C20-烷基)、芳基烯基、芳基炔基或杂芳基烷基(例如,苄基)。在一种实施方式中,Q为烷基或苄基;
R2和R2a独立地为氢、烷基、芳基、芳基烷基、杂芳基、羟基、氨基或烷氧基(优选氢),或者R2和R2a共同为=O;
R选自氢或者取代或未取代的、饱和或不饱和的脂肪族或芳香族基团,包括低级烷基、链烯基、芳基、杂环基、苄基、羟烷基和-CH2R3;
R3选自氢、低级烷基、链烯基、芳基、杂环基、苄基和羟烷基,包括环烷基和乙烯基;
R4选自氢、羟基、氨基、低级烷氧基或者取代或未取代的、饱和或不饱和的脂肪族或芳香族基团,包括C1-C20烷基和被羟基或羰基(例如,氧代)取代的C1-C20烷基,优选氢和3-氧代-5-环戊基-1-戊基;
R5为氢或者取代或未取代的低级烷基,优选未取代的低级烷基,优选甲基或乙基;
R6为取代或未取代的低级烷基,优选未取代的低级烷基,优选甲基或乙基:
R7选自氢、羟基、氨基、低级烷氧基或者取代或未取代的、饱和或不饱和的脂肪族或芳香族基团,优选氢或羟基;或者G、R4、R5、R6和/或R7共同可以形成1-3个或更多个环,所述环具有任选的额外的取代基,和/或
Q和R3共同可以形成1-3个或更多个环,所述环具有任选的额外的取代基,和/或
Q和R2共同可以形成1-3个或更多个环,所述环具有任选的额外的取代基。
前述结构的子集包括:
II.以上所示结构的2,6-亚甲基-3-苯并吖辛因,其中,R4、R5、R6和R7不形成额外的环;
其中,G、Q、Y、R2、R2a、R3、R4、R5、R6和/或R7如上所定义。
优选,R3选自氢、环丙基、环丁基、苯基、乙烯基、二甲基乙烯基、羟基环丙基、呋喃基和四氢呋喃基;
R4选自氢、羟基、氨基、低级烷氧基、C1-C20烷基和被羟基或羰基取代的C1-C20烷基;
R5为低级烷基;
R6为低级烷基;和
R7选自氢和羟基。
IIIa.吗啡喃,其中,R5和R6形成环,且R7为氢:
其中,G、Q、Y、R2、R2a、R3和R4如上所定义,且R19、R20、R21、R22和R23为氢或者取代或未取代的脂肪族或芳香族基团,或者共同可以形成杂环或碳环。
优选,R19为氢或低级烷基;
R20选自氢、低级烷基和低级羟烷基;
R21为氢;
R22选自氢、羟基、低级烷氧基和-NR13R14;或者
R21和R22共同形成羰基(=O)或乙烯基取代基(=CH2);
或者,R4和R21共同形成环;
R13和R14独立地选自氢、C1-C6烷基和C1-C6酰基;和
R23为氢、烷基(例如甲基),或者R19和R23共同形成第二个键。
IIIb.吗啡喃,其中,R5和R6形成环,且R7为羟基:
其中,G、Q、Y、R2、R2a、R3、R4、R19、R20、R21、R22和R23如上所定义。
IV.吗啡喃,其中,R5、R6和R7形成两个环:
其中,G、Q、Y、R2、R2a、R3、R4、R19、R20、R21、R22和R23如上所定义。
V.吗啡喃,其中,R4和R11形成额外的第六个环,该环可以为饱和或不饱和的(但不完全为芳香族的):
其中、G、Q、Y、R2、R2a、R3、R4、R19、R20、R21、R22和R23如上所定义。
除了主要的子集以外,存在本领域技术人员认识到与这些主要的子集密切相关的,但是不符合通常的马库什结构中的简单描述的化合物,例如,其中一个或多个R基团(例如,R4、R5、R6和/或R7)具有上述化学式中的一种的基团的化合物,从而形成二聚或杂二聚化合物。同样,“取代基”或通过两种或更多种变体形成的环预期包括这些二聚和杂二聚化合物。作为一种选择或除此以外,一个或多个R基团的取代基可以为取代的吗啡喃基,例如纳曲酮、甲基-纳曲酮或选自以下图表1-3的基团等。
在另一方面,本发明涉及与前述相关的化合物,其中,G具有下式:
这些化合物具有下式:
其中,
为1-3个或更多个环的芳基或杂芳基残基,和
A为化学键或连接基,例如(CH2)n,其中,一个或多个CH2可以被替换为-O-、环烷基或-CR1aR1b;
R1a和R1b独立地选自氢、卤素、低级烷基、低级烷氧基和低级烷硫基;
R10为氢或者一个或两个取代基,包括,例如,独立地选自氢、羟基、卤素、(C1-C6)烷基、(C1-C6)烷氧基、(C1-C6)卤代烷基和(C1-C6)卤代烷氧基和(C1-C6)烷硫基的残基;
R11为H或
为1-3个环的芳基或杂芳基残基;
A’为连接基,例如(CH2)m,其中,一个或多个CH2可被替换为-O-、环烷基、-CR1aR1b、-C(=O)-或-NH-;
R12选自氢和低级烷基;
R15为一个或两个独立地选自氢、羟基、卤素、(C1-C6)烷基、(C1-C6)烷氧基、(C1-C6)卤代烷基和(C1-C6)卤代烷氧基和(C1-C6)烷硫基的残基;
m为0或1-6的整数;和
n为1-6的整数;和
Q、Y、R2、R2a、R3、R4、R5、R6和R7如上所定义。
优选,Q选自烷基和苄基。
在一种实施方式中,A不是CH2。
前述结构(VI)的子集包括与以上所列的平行的种类,例如其中R4、R5、R6和R7不形成额外的环的2,6-亚甲基-3-苯并吖辛因;其中R5和R6形成一个环的吗啡喃;其中R5、R6和R7形成两个环的吗啡喃;和其中R4和R21形成可以为饱和或不饱和的第六个环的吗啡喃,等。
附图说明
图1为腹泻百分比与剂量的关系图,说明用化合物6治疗的小鼠对PGE2-诱发的腹泻的吗啡阻滞的抑制(腹膜内和口服给药)。
图2为反应潜伏期(单位:秒)与剂量的关系图,说明在甩尾测试中化合物6对吗啡-诱导的痛觉缺失的影响(腹膜内和口服给药)。
图3为反应潜伏期(单位:秒)与剂量的关系图,说明在热板测试中化合物6对吗啡-诱导的痛觉缺失的影响(腹膜内和口服给药)。
图4为剂量与时间(单位:分钟)的关系图,说明在各种给药形式中化合物6的药物动力学结果。
图5为腹泻百分比与剂量的关系图,说明用化合物12治疗的小鼠对PGE2-诱发的腹泻的吗啡阻滞的抑制(腹膜内和口服给药)。
图6为腹泻百分比与剂量的关系图,说明用化合物14治疗的小鼠PGE2-诱发的腹泻的吗啡阻滞的抑制(腹膜内和口服给药)。
图7为反应潜伏期(单位:秒)与剂量的关系图,说明在甩尾测试中化合物12对吗啡-诱导的痛觉缺失的影响(腹膜内和口服给药)。
图8为反应潜伏期(单位:秒)与剂量的关系图,说明在甩尾测试中化合物14对吗啡-诱导的痛觉缺失的影响(腹膜内和口服给药)。
图9为反应潜伏期(单位:秒)与剂量的关系图,说明在热板测试中化合物12对吗啡-诱导的痛觉缺失的影响(腹膜内和口服给药)。
图10为反应潜伏期(单位:秒)与剂量的关系图,说明在热板测试中化合物14对吗啡-诱导的痛觉缺失的影响(腹膜内和口服给药)。
具体实施方式
苯并吗吩烷和吗啡喃衍生物的酚羟基可以通过在美国专利6,784,187和7,057,035以及美国专利申请公开US 2007/0021457 A1中所述的简单、灵活和方便的路线,以化学方法转化为酰胺,这些专利均在此引入作为参考。
在一方面,本发明涉及具有下式的化合物以及这些化合物的各种衍生物或变体:
在另一方面,本发明涉及有下式的化合物:
在一个主要的子集中,R11为 且该化合物为具有下式的联苯、二芳基醚等:
在本领域中已知的是,μ、δ和κ激动剂的化合物表现出镇痛活性;选择性μ激动剂的化合物表现出止泻活性且可用于治疗运动障碍;μ拮抗剂和κ激动剂可用于治疗海洛因、精神刺激剂(即,可卡因、苯丙胺)、酒精和尼古丁上瘾;κ激动剂还是止痒剂且可用于治疗痛觉过敏。近来已经发现,κ激动剂也可用于治疗逆转录病毒感染(retroviral infection)[Peterson等,Biochem.Pharmacol.61,1141-1151(2001)]。通常情况下,以上类型III的吗啡喃的右旋异构体可用作镇咳药和抗惊厥药。
具有已知的高亲和力的阿片样物质受体配体示于以下图表中。在这些化合物中用G替换OH可产生表现出类似活性和更好生物利用度的化合物。本发明还包括以下G-取代的化合物的季铵化作用。
图表1阿片样物质受体配体
苯并吗啡喃(也称为2,6-亚甲基-3-苯并吖辛因)
环佐辛,R3=CH2-c-CH3H5 酮基环佐辛 乙基酮基环佐辛(EKC)
美他佐辛(metazocine),R3=CH3
非那佐辛,R3=CH2C6H5
SKF 10,047,R3=CH2CH=CH2
戊唑辛,R3=CH2CH=C(CH3)2
(均为外消旋)
MR2034-“Merz”核结构 MR2266 布马佐辛(bremazocine)
(旋光活性)
WIN 44,441
图表2阿片样物质受体配体
吗啡和吗啡喃
吗啡 纳曲酮,R17=CH2-c-CH3H5
纳洛酮,R17=CH2CH=CH2
纳美酮,R17=CH2CH=C(CH3)2
羟吗啡酮,R17=CH3
丁丙诺啡(buprenorphine) 二丙诺啡(diprenorphine)
埃托啡(N-Me;n-Pr vs Me)
纳洛芬 纳曲吲哚 纳布啡
β-纳曲胺 纳美芬 甲基纳曲酮
图表2(续)阿片样物质受体配体
吗啡和吗啡喃
SIOM(δ激动剂) nor-BNI(诺比那托费米(Norbinaltorphimine))
登记号=105618-26-6
左啡诺(Levorphanol),R17=CH3 右美沙芬(Dextromethorphan);R=CH3
赛克罗酚(Cyclorphan),R17=CH2-c-CH3H5 右啡烷(Dextrorphan);R=H
MCL 101,R17=CH2-c-C4H7 (注“反向”立体化学)
布托啡诺(Butorphanol),R17=CH2-c-C4H7和14-OH
Merz-吗啡喃杂化核,R17=CH2-(S)-四氢呋喃基
图表3混杂阿片样物质受体配体
登记号216531-48-5 登记号155836-52-5
登记号361444-66-8 R=CH3;登记号:69926-34-7
R=CH2CH2CH(OH)C6H11
登记号:119193-09-8
R=CH2CH(CH2Ph)CONHCH2CO2H;
登记号:156130-44-8
R=(CH2)3CH(CH3)2;登记号:151022-07-0
R=(CH2)3-2-噻吩基;登记号:149710-80-5
美普他酚(Meptazinol) 凯托米酮(Ketobemidone)
登记号59263-76-2 登记号469-79-4
登记号177284-71-8 曲马多活性代谢物
登记号80456-81-1
登记号189263-70-5 登记号173398-79-3
登记号189016-07-7 登记号189015-08-5
其它阿片样物质受体配体描述于Aldrich,J.V.《Burger′s MedicinalChemistry and Drug Discovery》中的“Analgesics(镇痛剂)”,M.E.Wolff编辑,John Wiley&Sons 1996,第321-44页,其公开的内容在此引入作为参考。在除两种以外的所有前述化合物中,根据本发明有一个酚OH被替换为G。在诺比那托费米和361444-66-8中有两个酚OH,其中之一或二者被替换为G。同样,氨基氮其中之一或二者可被季铵化。因此,本发明包括对含有一个或两个羟基部分和氨基氮的阿片配体改性的方法以及通过该方法生产的产品,所述方法包括用酰胺基或其它极性中性基团替换一个或多个羟基,然后使所述氨基氮季铵化。
本发明化合物可用于阻滞或逆转阿片剂的非CNS介导的副作用。待改善的一种具体副作用为抑制胃肠的运动。
定义
在整个说明书中,术语和取代基保持其定义不变。
烷基预期包括直链、支链或环状烃结构及其组合。组合例如为环丙基甲基。烃是指由氢和碳作为仅有的元素组成而组成的任何取代基。低级烷基是指具有1-6个碳原子的烷基。低级烷基的实例包括甲基、乙基、丙基、异丙基、环丙基、丁基、仲丁基和叔丁基、环丁基等。优选的烷基为C20或低于C20的烷基。环烷基为烷基的下位概念且包括具有3-8个或更多个碳原子的环状烃基。环烷基的实例包括环丙基、环丁基、环戊基、降冰片基(norbornyl)等。
链烯基是指含有至少一个双键的不饱和的无环烃基。这种基团含有2-10个或更多个碳原子,优选2-8个碳原子,更优选2-6个碳原子。合适的链烯基的实例包括丙烯基、丁烯-1-基、异丁烯基、戊烯-1-基、2-甲基丁烯-1-基、3-甲基丁烯-1-基、己烯-1-基、庚烯-1-基和辛烯-1-基、链二烯等。
炔基是指含有至少一个叁键的不饱和的无环烃基。这种基团含有2-10个或更多个碳原子,优选2-8个碳原子,更优选2-6个碳原子。合适的炔基的实例包括丙炔基、丁炔-1-基、戊炔-1-基、丁炔-2-基、3-甲基丁炔-1-基、己炔-1-基、庚炔-1-基和辛炔-1-基等。
环烷基或环烯基是指在具有3-10个碳原子,优选3-6个或更多个碳原子的环(或稠环体系)中的脂环族基团。合适的脂环族基团的实例包括环丙基、环丙烯基、环丁基、环戊基、环己基、环己烯基等。
氧杂烷基(oxaalkyl)是指其中一个或多个碳原子被替换为氧的烷基残基。其实例包括甲氧基丙氧基、3,6,9-三氧杂癸基(trioxadecyl)等。
烷氧基(alkoxy)或烷氧基(alkoxyl)是指通过氧原子与母体结构连接的具有1-8个或更多个碳原子的直链、支链、环状构型的基团及其组合。其实例包括甲氧基、乙氧基、丙氧基、异丙氧基、环丙氧基、环己氧基等。低级烷氧基是指含有1-4个碳原子的基团。
酰基是指通过羰基官能团与母体结构连接的甲酰基和具有1、2、3、4、5、6、7和8个或更多个碳原子的直链、支链、环状构型、饱和的、不饱和的和芳族基团及其组合。酰基残基中的一个或多个碳原子可以被替换为氮、氧或硫,或者两个氢原子可以被氧替换或间隔,只要与母体的连接点保持在羰基上即可。其实例包括乙酰基、苯甲酰基、丙酰基、异丁酰基、叔丁氧基羰基、苄氧基羰基等。低级酰基是指含有1-6个或更多个(例如4个)碳原子的基团。
芳基和杂芳基是指含有0-3个选自O、N或S的杂原子的5元或6元芳环或杂芳环;含有0-3个选自O、N或S的杂原子的双环9元或10元芳环或杂芳环体系;或者含有0-3个选自O、N或S的杂原子的三环13元或14元芳环或杂芳环体系。芳香族6元至14元碳环包括,例如,苯、萘、l,2-二氢化茚、四氢化萘和芴,5元至10元芳香族杂环包括,例如,咪唑、吡啶、吲哚、噻吩、噻唑、呋喃、苯并咪唑、喹啉、异喹啉、喹喔啉、嘧啶、吡嗪、四唑和吡唑。
芳基烷基或芳烷基是指与芳环连接的烷基残基。其实例为苄基、苯乙基等。杂芳基烷基是指与杂芳环连接的烷基残基。其实例包括,例如,吡啶基甲基、嘧啶基乙基等。
杂环是指其中一个或两个或更多个碳原子被替换为杂原子(例如氧、氮或硫)的环烷基或芳基残基。杂芳基构成杂环的下位概念。落入本发明范围内的杂环的实例包括吡咯烷、吡唑、吡咯、吲哚、喹啉、异喹啉、四氢异喹啉、苯并呋喃、苯并二氧六环(benzodioxan)、苯并恶烷(benzodioxole)(当作为取代基出现时,通常称为亚甲基二氧基苯基)、四唑、吗啉、噻唑、吡啶、哒嗪、嘧啶、噻吩、呋喃、噁唑、噁唑啉、异噁唑、二氧杂环乙烷、四氢呋喃等。
取代的烷基、芳基、环烷基、杂环基等是指其中各残基上高达3个或更多个H原子被替换为以下基团的烷基、芳基、环烷基或杂环基,所述基团例如卤素、卤代烷基、烷基、酰基、烷氧基烷基、低级羟烷基、苯基、杂芳基、苯磺酰基、羟基、低级烷氧基、卤代烷氧基、羧基、烷氧羰基(也称为烷氧基羰基)、烷氧基羰基氨基、酰胺基(也称为烷基氨基羰基)、氰基、羰基(也称为氧代)、乙酰氧基、硝基、氨基、烷基氨基、二烷基氨基、巯基、烷硫基、亚砜、砜、磺酰基氨基、酰氨基、脒基、芳基、苄基、杂环基、苯氧基、苄氧基、杂芳氧基、羟基亚氨基、烷氧基亚氨基、氧杂烷基、氨基磺酰基、三苯甲基、脒基、胍基、脲基和苄氧基。
术语“药学上可接受的盐”是指其抗衡离子衍生自药学上可接受的无毒酸和碱的盐。用于本发明化合物的合适的药学上可接受的碱加成盐包括无机酸、有机酸,且由于该化合物具有季铵基团,该药学上可接受的碱加成盐还包括水(其在形式上以氢氧根阴离子提供)。抗衡离子的实例包括氢氧化物、乙酸盐、苯磺酸盐、苯甲酸盐、碳酸氢盐、硫酸氢盐、碳酸盐、樟脑磺酸盐(camphorsulfonate)、柠檬酸盐、乙磺酸盐、反丁烯二酸盐、葡糖酸盐、谷氨酸盐、乙醇酸盐、溴化物、盐酸盐、羟乙磺酸盐(isethionate)、乳酸盐、马来酸盐、苹果酸盐、扁桃酸盐、甲磺酸盐(methanesulfonate)、粘酸盐(mucate)、硝酸盐、扑酸盐(pamoate)、泛酸盐、磷酸盐、琥珀酸盐、硫酸盐、酒石酸盐、三氟醋酸盐、对甲苯磺酸盐、乙酰苯甲酸甲酯(acetamidobenzoate)、己二酸盐、藻酸盐、氨基水杨酸盐、脱水亚甲基柠檬酸盐(anhydromethylenecitrate)、抗坏血酸盐、天冬氨酸盐、乙二胺四乙酸钙、樟脑酸盐(camphorate)、樟脑磺酸盐(camsylate)、癸酸盐、己酸盐、辛酸盐、肉桂酸盐、环磺酸盐、二氯乙酸盐、乙二胺四乙酸盐(EDTA)、乙二磺酸盐(edisylate)、双羟萘酸盐(embonate)、丙酸酯月桂硫酸酯(estolate)、乙磺酸盐(esylate)、氟化物、甲酸盐、龙胆酸盐、葡庚糖酸盐(gluceptate)、葡糖醛酸盐、甘油磷酸盐、乙醇酸盐、对羟乙酰氨基苯胂酸盐(glycollylarsanilate)、己基间苯二酚盐(hexylresorcinate)、马尿酸盐、羟基萘甲酸盐(hydroxynaphthoate)、碘化物、乳糖醛酸盐(lactobionate)、丙二酸盐、甲磺酸盐、萘二磺酸盐(napadisylate)、萘磺酸盐、烟酸盐、油酸盐、乳清酸盐(orotate)、草酸盐、酮戊二酸盐(oxoglutarate)、棕榈酸盐、果胶酸盐(pectinate)、果胶酸盐聚合物、苯巴比妥盐(phenylethylbarbiturate)、苦味酸盐、氧脯氨酸盐(pidolate)、丙酸盐、硫氰酸盐(rhodanide)、水杨酸盐、癸二酸盐、硬脂酸盐、丹宁酸盐、8-氯茶碱(theoclate)、甲苯磺酸盐(tosylate)等。
当化合物含有酸性残基时,该化合物可作为两性离子存在。用于本发明化合物的其它合适的药学上可接受的碱加成盐包括铵、由铝、钙、锂、镁、钾、钠和锌制成的金属盐、或由赖氨酸、N,N′-二苄基乙二胺、氯普鲁卡因、胆碱、二乙醇胺、乙二胺、甲葡胺(N-甲基葡糖胺)和普鲁卡因制成的有机盐。其它碱加成盐包括由以下物质制成的盐:槟榔碱、精氨酸、钡、苯乙苄胺(benethamine)、苄星、甜菜碱、铋、克立咪唑(clemizole)、铜、二甲基乙醇胺、二乙胺、二乙氨基乙醇(diethylaminoethanol)、吡咯乙醇(epolamine)、乙二胺、铁、亚铁、葡糖胺(glucamine)、氨基葡萄糖(glucosamine)、组氨酸、海巴明(hydrabamine)、咪唑、异丙胺、锰、亚锰、甲基葡糖胺(methylglucamine)、吗啉、4-羟乙基吗啉(morpholineethanol)、N-乙基吗啉、N-乙基哌啶、哌嗪、哌啶、多元胺树脂、嘌呤、可可碱、三乙胺、三甲胺、三丙胺、三乙醇胺和氨基丁三醇(tromethamine)。
本发明的化合物为盐,因此具有抗衡离子。本发明的抗衡离子为药学上可接受的抗衡离子。药学上可接受的抗衡离子包括,例如,卤化物、硫酸盐、磷酸盐、硝酸盐和阴离子有机化合物。卤化物包括碘化物、溴化物、氯化物及其组合。
本领域技术人员应理解,如果本发明的化合物制备成一种盐(例如碘化物盐),则使其通过阴离子交换柱可以容易地将所述盐转化为另一种盐(例如溴化物盐)。
事实上,本文所述的所有化合物都含有一个或多个不对称中心,因此可以产生对映异构体、非对映异构体、以及可根据绝对立体化学定义为如(R)-或(S)-的其它立体异构的形式。本发明意欲包括所有这些可能的异构体以及它们的外消旋和旋光纯形式。通常,已发现,吗啡喃和苯并吗吩烷的左旋异构体为更有效的抗伤害性疼痛的药物,而其右旋异构体可用作镇咳药或解痉药。旋光活性(R)-和(S)-异构体可以用手性合成单体(synthon)或手性试剂制得,或者用常规的方法进行拆分。当本文所述的化合物含有烯属双键或其它几何不对称中心时,除非另外说明,所述化合物意欲包括E和Z几何异构体二者。同样,还意欲包括所有的互变异构体形式。
任意地描述在氮上新产生的手性中心的构型。在一些情况下,该描述可以指R构型,而在一些情况下,该描述可以指S构型。这些描述不应看作是说明绝对立体化学已被确定。应当理解的是,以形成手性分子的氮的烷基化(大多数)可能优选一种异构体。在其中已确定手性的一些情况下,在N处R构型占绝大多数,且重结晶时回收的非对映异构体在N处具有R构型。人们假定,还可形成一些有限量的相对的构型,且可保留在母液中。如果需要,通过本领域技术人员众所周知的方法可以回收或产生该异构体和/或任何外消旋或非对映异构的混合物。此外,还可能存在几个手性碳原子。除非在权利要求中明确说明,所述权利要求意欲包括二种或所有异构体及混合物,而无论该权利要求是否采用说明中心手性的化学式。
缩写
有机化学者(即本领域普通技术人员)采用的缩写的全面列举出现在Journal of Organic Chemistry各卷的第一期。通常在题为“缩写的标准列举(Standard List of Abbreviations)”的表中出现的列举在此引入作为参考。以下缩写和术语在全文中具有所示含义:
Ac=乙酰基
BNB=4-溴甲基-3-硝基苯甲酸
Boc=叔丁氧基羰基
Bu=丁基
c-=环
CHO=中国仓鼠卵巢
DAMGO=络氨酸酶-丙氨酰甘氨酸-NMePhe-NHCH2OH(Tyr-ala-Gly-NMePhe-NHCH2OH)
DBU=二氮杂二环[5.4.0]十一-7-烯
DCM=二氯甲烷=甲叉二氯=CH2Cl2
DEAD=氮氮二甲酸二乙酯(diethyl azodicarboxylate)
DIC=二异丙基碳二亚胺(diisopropylcarbodiimide)
DIEA=N,N-二异丙基乙胺(N,N-diisopropylethyl amine)
DMAP=4-N,N-二甲氨基吡啶(4-N,N-dimethylaminopyridine)
DMF=N,N-二甲基甲酰胺(N,N-dimethylformamide)
DMSO=二甲基亚砜
DOR=δ阿片样物质受体
DPPF=1,1′-双(二苯膦基)二茂铁
DVB=l,4-二乙烯基苯
EC50=产生50%效应的药物浓度
EEDQ=2-乙氧基-1-乙氧基羰基-1,2-二氢喹啉
EGTA=乙二醇四乙酸(ethylene glycol tetraacetic acid)
Emax=(药物的)最大效应
Fmoc=9-芴甲氧羰基(9-fluorenylmethoxycarbonyl)
GC=气相色谱
GI=胃肠
HATU=O-(7-氮杂苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐
HOAc=乙酸
HOBt=羟基苯并三唑
IC50=产生50%抑制的药物浓度
Imax(药物的)最大抑制
IP=腹膜内
IV=静脉内
KOR=κ阿片样物质受体
Me=甲基
mesyl=甲磺酰基
mNTX=甲基-纳曲酮
MOR=μ阿片样物质受体
MRL=最大反应潜伏期
MTBE=甲基叔丁基醚
NMO=N-甲基吗啉氧化物
NOESY=核欧沃豪增强光谱
PD=药效
PEG=聚乙二醇
PGE2=前列腺素E2
Ph=苯基
PhOH=苯酚
PfP=五氟苯酚
PK=药物动力学
PO=口服给药
PPTS=对甲基苯磺酸吡啶
PyBroP=溴-三-吡咯烷基-六氟磷酸膦
(bromo-tris-pyrrolidino-phosphonium hexafluorophosphate)
rt室温
sat’d=饱和
s-=仲
SC=皮下
t-=叔
TBDMS=叔丁基二甲基甲硅烷基(t-butyldimethylsilyl)
TFA=三氟乙酸
THF=四氢呋喃
TMOF=原甲酸三甲酯
TMS=三甲基甲硅烷基(trimethylsilyl)
tosyl=对甲苯磺酰基(p-toluenesulfonyl)
Trt=三苯基甲基
U50,488=κ激动剂
在将苯酚转化为所需的生物立体(biostere)或季铵化的过程中,在所研究的物质中的残基可能需要保护和去保护。与“保护”、“去保护”和“受保护”官能团相关的术语出现在整个申请中。这样的术语应被本领域技术人员充分理解,并在涉及使用一系列试剂进行连续处理的方法的上下文中使用。在上下文中,保护基是指在处理步骤期间掩蔽官能团的基团,该官能团将会另外发生反应,而该反应是不期望的。所述保护基防止在那个步骤中的反应,但是随后可以将其除去从而使最初的官能团暴露。所述除去或“去保护”发生在所述官能团将受到干扰的那步或几步反应完成之后。因此,当试剂的顺序在下文中得到详细说明时,本领域普通技术人员能容易预见那些适于作为“保护基”的基团。用于该目的的合适的基团已在化学领域的标准教科书中作了详细的描述,例如T.W.Greene的Protective Groups in OyganicSynthesis(有机合成中的保护基)[John Wiley&Sons,New York,1991],其在此引入作为参考。
本发明的优选的化合物通过用适当的烷基化试剂(例如,甲基卤化物或硫酸盐)使叔胺前体季铵化而制备:
反应式1
用于制备本发明化合物的起始化合物可通过在美国专利6,784,187和7,057,035以及美国专利申请公开US 2007/0021457中所述的路线中的一种来制备。例如:
反应式2
反应式3
反应式4
反应式2中所示的N-羟基琥珀酰亚胺酯中间体(3)可通过美国专利7,057,035的方法来制备,该专利在此引入作为参考。如下所述,使所述N-羟基琥珀酰亚胺酯随后与适当的芳基烷基胺(4)反应。作为一种选择,采用在反应式3中所示的直接羰基化/酰胺化。如反应式4所示,许多二芳基化合物可通过Suzuki偶联来制备。
实验部分
用实例化合物进行体外和体内研究。体外研究定义这些分子的受体结合和功能活性。进行体内研究以说明外周至中枢神经系统的相对活性。对于一个实例(化合物6),进行药效学研究以说明PGE2-诱发的腹泻的吗啡阻滞的抑制能力。
阿片样物质受体结合测定
本发明的发明人已经检验了该系列化合物的阿片样物质受体结合亲和力。用于筛分化合物的结合测定与Neumeyer等人以前在Design and Synthesisof Novel Dimer Morphinan Ligands for κand μOpioid Receptors.(用于κ和μ阿片样物质受体的新型二聚吗啡喃配体的设计和合成)J.Med.Chem.2003,46,5162中报道的那些类似。在1nM[3H]U69,593(κ)、0.25nM[3H]DAMGO(μ)或0.2nM[3H]纳曲吲哚(δ)存在下,在最终体积为1mL的50mM Tris-HCl(pH 7.5)中,于25℃下,来自稳定地表达一种类型的人类阿片样物质受体的CHO细胞的膜蛋白用12种不同浓度的化合物培养。[3H]U69,593和[3H]DAMGO的培养时间为60分钟。由于[3H]纳曲吲哚与受体缔合较慢,该放射配体的培养时间为3小时。用[3H]纳曲吲哚培养的样品还含有10mMMgCl2和0.5mM苯甲磺酰氟(phenylmethylsulfonyl fluoride)。通过包含10μM纳洛酮来测量非特异性结合。使用Brandel 48-孔细胞采集器,通过使样品通过Schleicher&Schuell 32号玻璃纤维过滤器进行过滤以终止所述结合。该过滤器随后用3mL冷的50mM Tris-HCl(pH 7.5)洗涤3次,并在2mLEcoscint A闪烁流体中计数。对于[3H]纳曲吲哚和[3H]U69,593结合,在使用前,使所述过滤器在0.1%聚乙烯亚胺中浸泡至少60分钟。通过适于对数-概率单位分析的最小平方来计算IC50值。未标记的化合物的Ki值由方程式Ki=(IC50)/1+S来计算,其中S=(放射配体的浓度)/(放射配体的Kd)。数据为来自一式三份进行的至少三个实验的平均值±标准误差的平均值(SEM)。
[35S]GTPγS结合测定
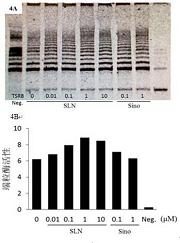
用于筛分化合物的测定与Wentland等人以前在“Redefining thestructure-activity relationships of 2,6-methano-3-benzazocines Part 4.Opioidreceptor binding properties of 8-[N-(4′-phenyl)-phenethyl)carboxamido]analogues of cyclazocine and EKC(重新定义2,6-亚甲基-3-苯并吖辛因的结构-活性关系,第4部分,环佐辛和EKC的8-[N-(4′-苯基)-苯乙基)酰胺基]类似物的阿片样物质受体结合性能)”J.Med.Chem.2006,49,5635中报道的那些类似。在最终体积为0.5mL中,12种不同浓度的各测试化合物用稳定表达人类κ或μ阿片样物质受体的15μg(κ)、10μg(δ)或7.5μg(μ)的CHO细胞膜培养。该测定缓冲液由50mM Tris-HCl(pH 7.4)、3mM MgCl2、0.2mMEGTA、3μM GDP和100mM NaCl组成。[35S]GTPγS的最终浓度为0.080nM。通过包含10μM GTPγS来测量非特异性结合。通过加入膜引发结合。于30℃下培养60分钟后,使样品通过Schleicher&Schuell 32号玻璃纤维过滤器进行过滤。该过滤器用冷的50mM Tris-HCl(pH 7.5)洗涤3次,并在2mL的Ecoscint闪烁流体中计数。数据为来自一式三份进行的至少三个单独的实验的平均Emax和EC50值±标准误差的平均值(S.E.M)。为了计算Emax值,基部的[35S]GTPγS结合设定为0%。为了确定化合物在所述μ阿片样物质受体上的拮抗活性,在200nM μ激动剂DAMGO存在下,将表达μ阿片样物质受体的CHO膜用12种不同浓度的化合物培养。为了确定化合物在κ阿片样物质受体上的拮抗剂活性,在100nM κ激动剂U50,488存在下,将表达κ阿片样物质受体的CHO膜用所述化合物培养。为了确定化合物在δ受体上是否为拮抗剂,在10nM δ-选择性激动剂SNC 80存在下,将表达δ受体的CHO膜用12种不同浓度的测试化合物培养。
通过Jiang等人在[J.Pharmacol.Exp.Ther.264,1021-1027(1993),第1022页]中所述的方法来评价其副作用待改善的阿片剂的抗伤害性疼痛的活性(antinociceptive activity)。
通过Gmerek,Debra E.;Cowan,Alan;Woods,James H.在“Independent central and peripheral mediation of morphine-induced inhibition ofgastrointestinal transit in rats(老鼠胃肠输送的吗啡-诱导的抑制的独立中枢神经系统和外周调解)”Journal of Pharmacology and Experimental Therapeutics(1986),236(1),8-13中所述的方法来评价对胃肠运动的影响。
肠运动的PGE2模式
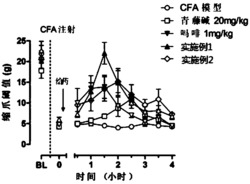
为了评定新型外周作用阿片拮抗剂的效果,使用肠运动的PGE2(一种前列腺素)模式。在小鼠中腹膜内(IP)注射(0.1mg/kg)的15分钟内PGE2诱发腹泻。用吗啡(1mg/kg)预治疗(30分钟)阻断该效应。测试外周作用阿片拮抗剂对吗啡的腹泻阻滞的抑制能力。
每种阿片拮抗剂用吗啡测试,以建立吗啡阻断PGE2-诱发的腹泻能力的剂量-反应拮抗。对小鼠(n=10只/组)腹膜内注射或口服(PO)给药(通过管饲法)新型阿片拮抗剂(0-3mg/kg,IP;0-30mg/kg,PO),并放置在树脂玻璃饼式笼(Braintree Scientific,Braintree,MA)中。每个饼式笼装有多达10只小鼠,且直径为21.5cm,高7.5cm。各室为5cm(底部)×9cm(长)。为了最初的研究,在给药新型阿片拮抗剂15分钟后给予吗啡(1mg/kg,IP),并让小鼠返回所述饼式笼。30分钟后,用PGE2(0.1mg/kg,IP)治疗小鼠,并让小鼠再次返回饼式笼。在最终观察期间,在PGE2给药15分钟后记录是否存在腹泻。小鼠仅测试一次。盐水-盐水(10mL/kg,IP)和盐水-吗啡为阳性对照组,所有结果与盐水-吗啡治疗组相比较。数据代表在最终观察时患有PGE2-诱发的腹泻的小鼠的百分比。
抗伤害性疼痛的甩尾测试
使用市售可得的甩尾装置(Columbus Instruments,Columbus,OH)评定对剧烈热刺激的伤害性疼痛的抗性。甩尾测试的主旨主要是外周反射反应测定。在该标准模式中,使小鼠轻微受约束,且将其尾部放置在热光束上。一旦光束开启(立即开启;9.3瓦),记录反射性轻弹所述尾部所需的时间。最大反应潜伏期(MRL)设定为10秒,以避免与较长暴露时间相关的潜在的热伤害。如果在10秒后无反应,将小鼠移开,并记录最大反应潜伏期(MRL;10秒)。
吗啡(15mg/kg,IP,在测试前45分钟给药)产生MRL或接近MRL。每种阿片拮抗剂用吗啡测试,以建立在甩尾测试中吗啡-诱导的抗伤害性疼痛的剂量-反应拮抗。小鼠(n=10只/组)首先在甩尾测试中测试,以确定基线反应。如果基线反应时间大于10秒,则将该小鼠排除于研究之外。对小鼠给药不同剂量的阿片拮抗剂(IP或PO,在甩尾测试的测试前60分钟)。15分钟后,注射吗啡(IP,15mg/kg,在甩尾测试的测试前45分钟)。所有结果与盐水-吗啡治疗组平均反应潜伏期相比较。
抗伤害性疼痛的热板测试
使用市售可得的热板装置(Columbus Instruments,Columbus,OH)评定对剧烈热刺激的伤害性疼痛的抗性。热板测试主旨为伤害性疼痛的脊椎测定。热板方法包括将每只家鼠放置在已加热的表面上,并激活定时器。将小鼠单独放置在热板(25.4cm×25.4em,由丙烯酸的盒子环绕,以防止动物逃脱)上;表面温度=55℃),记录舔任一只后爪的反应潜伏期。最大反应潜伏期(MRL)设定为60秒,以避免与较长暴露时间相关的潜在的热伤害。当其响应热而舔任一只后爪时或者在60秒过去后将小鼠从已加热的表面移开。记录反应潜伏期,使小鼠返回其家用笼。
吗啡(15mg/kg,IP,在测试前45分钟给药)产生MRL或接近MRL。每种阿片拮抗剂用吗啡测试,以建立吗啡-诱导的抗伤害性疼痛的剂量-反应拮抗。使小鼠(n=10只/组)首先测试,以确定基线反应。如果基线反应时间大于30秒,则将该小鼠排除于研究之外。随后将小鼠给药不同剂量的阿片拮抗剂(IP或PO,在热板测试的测试前60分钟)。15分钟后,注射吗啡(IP,15mg/kg,在热板测试的测试前45分钟)。所有结果与盐水-吗啡治疗组平均反应潜伏期相比较。
化合物6(如下所示)的PK评价方法。给动物服用所述实例化合物,使用以下方法将血液样品收集2小时。用1-2%的异氟烷将老鼠暂时麻痹,将来自外侧尾静脉的血液样品(总血量约250mL)收集至含有EDTA的管中。使所述管在10K x g下离心分离2分钟,以分离血浆。将血浆移液至微量离心瓶中,并于-80℃下储存,直至通过液相色谱法-质谱法/质谱法(LC-MS/MS)(Baranczewski等人,2006的修改)确定血浆含量。用于这些研究的定量下限(LOQ)为1.0ng/mL,且测定的偏差系数<4.4%。对于每个时间点,计算血浆中实例化合物的平均浓度。如果该值低于LOQ,则表示为该值为0。
开发生物分析测定并有资格测量老鼠血浆中的实例化合物。该方法包括通过高效液相色谱法联同PE/Sciex API 2000质谱仪(LC-MS/MS)来分析老鼠血浆的乙腈沉淀的蛋白提取物。通过用纳曲酮(Sigma Chemical,St.Louis,MO)掺加空白血浆来制备测定标准物和对照物,以实现标准物的浓度为100ng/mL,在每次样品分析过程中将其进一步稀释,使测定对照物的浓度为80ng/mL、40ng/mL和8ng/mL。通过将标准物、样品和对照物各100mL转移至含有10mL内标(在乙腈中的1mg/mL氢可酮(hydrocodone))和10mL 10mM碳酸氢钠缓冲液的微量离心管中进行萃取。随后使用250mL乙腈来沉淀蛋白,将澄清的上清液移除,浓缩至干,用100mL流动相缓冲液混合物重组。为了进行分析,将5mL重组的提取物注射在LC-MS/MS体系中。使用Waters C183.5m柱(XBridge,内径2.1×50mm,Milford,MA),于环境温度下均一浓度下(isocratically)实施高效液相色谱法。所述流动相由10mM乙酸铵、0.1%氢氧化铵缓冲液(pH 9.0+0.5)和乙腈(45∶55,v/v)组成。流动速率为0.350mL/分钟。将API2000(Applied Biosystems,Forest City,CA)三倍四极配备Turbo离子喷雾源。使用正离子模式测量纳曲酮产物离子m/z342→324的峰面积以及内标的产物离子m/z 300→199的峰面积。离子喷雾电压设定为4500V,喷雾器气体为25psi,加热器气体为55psi,探针温度为350℃。使用分析软件(Applied Biosystems,1.2版本)进行数据分析。绘制峰面积比(分析物/内标)与分析物标称浓度的标准曲线,其中权重因数为1/y。在1ng/mL至100ng/mL范围内标准曲线为线性的,推定系数(r2)>0.990(n=10)。纳曲酮的LOQ限定为1ng/mL。通过在同一天分析每种80ng/mL、40ng/mL和8ng/mL测定对照物(在每种浓度下,n=5)来评价当天的准确度和精确度。用测量浓度与标称浓度的百分比计算准确度,精确度用偏差系数表示。准确度分别为86%、100%和103%,精确度分别为3.6%、2.9%和1.9%。对于每种80ng/mL、40ng/mL和8ng/mL测定对照物,评价10次不同试验(对于每种对照样品,n=26)的当天准确度和精确度。准确度分别为103%、103%和115%,精确度分别为10.1%、14.1%和19.1%。在于-69℃下冷冻然后解冻的三次循环后,通过分析在80ng/mL、40ng/mL和8ng/mL浓度下的测定对照样品来确定冷冻和解冻稳定性。稳定性用测量浓度与所述标称浓度的百分比表示。循环1的%恢复为97%,循环2的%恢复为100%,循环3的%恢复为98%。
实验结果
在正文、表格和图中,化合物(cmpds)2、4、6、8、10、12和14是指以下化合物:
在表1中,较低数字说明化合物对该受体更有效。例如,化合物8对μ受体具有高的亲和力(Ki=0.91nM),但是对δ受体具有很低的亲和力(Ki=550nM)。
表1:结合亲和力-与纳曲酮相比较
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
![]()





动态评分
0.0