

IPC分类号 : C07F15/00I,B01J31/22I,C07B37/00I,C07C1/32I,C07C15/14I,C07C41/30I,C07C43/205I,C07C45/68I,C07C49/784I,C07C201/12I,C07C205/35I,C07C253/30I,C07C255/50I







专利摘要
本发明涉及一种三唑卡宾钯金属配合物及制备方法和应用,以N‑吡啶基三唑为分子骨架,以1,2,3‑三唑上碳原子和吡啶氮原子与金属钯配位所制备的一种三唑卡宾钯金属配合物,其化学式为C17H16Cl2N4O2Pd。该三唑卡宾钯金属配合物作为催化剂在氯代芳烃或者溴代芳烃与芳基硼酸的Suzuki偶联反应中的应用中具有选择率高、转化率高的特点。
权利要求
1.一种三唑卡宾钯金属配合物,其特征在于,结构式为:
2.一种如权利要求1所述的三唑卡宾钯金属配合物的制备方法,其特征在于,包括以下步骤:
(1)合成1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐
以二氯甲烷或甲醇为溶剂,将2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯与[Me
(2)合成三唑卡宾钯金属配合物
以乙腈为溶剂,将1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐、氧化银与Me
3.根据权利要求2所述的三唑卡宾钯金属配合物的制备方法,其特征在于,步骤(1)中2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯通过以下过程制得:
以甲苯为溶剂,将7-乙氧羰基吡啶并[1,5-a]四氮唑、苯乙炔、三氟甲磺酸铜按摩尔比为1:(1.5~2):(0.05~0.1)在氮气保护下于80~100℃条件下反应8~12小时,得到2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯。
4.根据权利要求3所述的三唑卡宾钯金属配合物的制备方法,其特征在于,1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐与Pd(CH
7-乙氧羰基吡啶并[1,5-a]四氮唑通过以下过程制得:
以甲苯为溶剂,将1-氧-4-乙氧羰基吡啶、对甲苯磺酰氯、叠氮化钠按摩尔比为1:(2~3):(1.5~3)于100~120℃条件下反应18~24小时,得到7-乙氧羰基吡啶并[1,5-a]四氮唑。
5.一种如权利要求2所述方法制备的三唑卡宾钯金属配合物的应用,其特征在于,氮气保护下,向反应容器中加入氯代芳烃、芳基硼酸、碱、三唑卡宾钯金属配合物与反应溶剂,或者向反应容器中加入溴代芳烃、芳基硼酸、碱、三唑卡宾钯金属配合物与反应溶剂,然后在20~120℃下反应2~24小时,反应完成后,经分离纯化得到Suzuki交叉偶联产物。
6.根据权利要求5所述的三唑卡宾钯金属配合物的应用,其特征在于,反应溶剂是乙醇、N,N-二甲基甲酰胺、异丙醇、二氧六环、甲醇与二甲基亚砜中的一种。
7.根据权利要求5所述的三唑卡宾钯金属配合物的应用,其特征在于,碱是碳酸钠、碳酸钾、碳酸铯、氢氧化钠、碳酸氢钾与叔丁醇钾中的一种。
8.根据权利要求5所述的三唑卡宾钯金属配合物的应用,其特征在于,氯代芳烃和碱的物质的量比、溴代芳烃和碱的物质的量比均为1:(1~3),氯代芳烃和芳基硼酸的物质的量比、溴代芳烃和芳基硼酸的物质的量比均为1:(1~2)。
9.根据权利要求5所述的三唑卡宾钯金属配合物的应用,其特征在于,所述的氯代芳烃与三唑卡宾钯金属配合物的物质的量比、溴代芳烃与三唑卡宾钯金属配合物的物质的量比均为1:(0.001~0.1)。
10.根据权利要求5所述的三唑卡宾钯金属配合物的应用,其特征在于,氯代芳烃或者溴代芳烃在反应溶剂中的浓度为0.08~0.16mol/L;芳基硼酸为苯基硼酸、4-甲基苯基硼酸、4-甲氧基苯基硼酸和3-硝基苯基硼酸中的一种。
说明书
技术领域
本发明涉及金属有机化学和催化技术领域,具体涉及一种三唑卡宾钯金属配合物及制备方法和应用。
背景技术
近年来过渡金属配合物的设计与合成发展迅速,过渡金属原子具有不饱和的d电子轨道,可以和供电子性的配体形成金属-配体化学键。氮杂环卡宾(NHCs)是一类具有强供电子能力的配体,目前是金属有机化学、过渡金属催化以及配位化学的研究热点之一,其中卡宾钯金属配合物在C–C键形成(Suzuki–Miyaura偶联、Heck–Mizoroki偶联、Sonogashira偶联)、 C–N键形成(Buchwald–Hartwig胺基化)以及不饱和键的催化氢化反应中具有良好的催化活性(E.A.B.Kantchev,et.al.Angew.Chem.Int.Ed.2007,46,2768;F.E.Hahn,et.al.Angew. Chem.Int.Ed.2008,47,3122;N.Marion,et.al.Acc.Chem.Res.2008,41,1440;S. Díez-González,et.al.Chem.Rev.2009,109,3612;D.Huang,et.al.Coord.Chem.Rev.2014,272, 145;P.Ruiz-Castillo,et.al.Chem.Rev.2016,116,12564)。与传统的膦配体相比,氮杂环卡宾配体具有毒性小、合成简单、供电子能力强、配位能力强等特点,而且该类配体以及制得的相应金属配合物通常对水、热和空气具有较好的稳定性,能更好地满足常规反应体系和工业放大的需要。
在以往的报道中,基于1,2,3-三唑的非正常/介离子氮杂卡宾(MIC)配合物备受关注,一方面是因为此类配体比正常的NHC配体有更强的σ供电子性质;另一方面该类配体可以通过经典的铜催化的“点击反应”(Click Reaction)来制备(J.M.Aizpurua,et.al.NewJ.Chem. 2014,38,474;B.Schulze,et.al.Chem.Soc.Rev.2014,43,2522)。由于不同骨架的氮杂环卡宾类金属配合物具有不同的结构参数和催化性能,因此,为了丰富现有的化合物种类,更好地探索和拓展该类配体的应用价值,有必要合成一种新型的吡啶基-1,2,3-三唑卡宾钯金属配合物,并将其作为催化剂应用于Suzuki偶联反应中制备联苯化合物(J.Huang,et.al.Eur.J.Org. Chem.2012,6630;S.Liu,et.al.J.Org.Chem.2014,79,3249)。联苯类化合物是一类非常重要的精细化学品和医药中间体,可以在过渡金属催化作用下,以价廉易得的氯代芳烃或者溴代芳烃与芳基硼酸为原料,通过Suzuki偶联反应一步合成,该方法具有操作简单、简洁高效、原子经济性高、环境友好等优点(A.Suzuki,et.al.Chem.Commun.2005,4759;A.J.J.Lennox, et.al.Chem.Soc.Rev.2014,43,412)。
发明内容
本发明所要解决的技术问题是提供一种三唑卡宾钯金属配合物。
本发明所要解决的另一个技术问题是提供该三唑卡宾钯金属配合物的制备方法。
本发明所要解决的第三个技术问题是提供该三唑卡宾钯金属配合物的应用。
为解决上述问题,本发明采用的技术方案如下:
一种三唑卡宾钯金属配合物,结构式为:
一种三唑卡宾钯金属配合物的制备方法,包括以下步骤:
(1)合成1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐
以二氯甲烷或甲醇为溶剂,将2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯与[Me3O]BF4按摩尔比为1:(1.1~1.5)在氮气保护下于20~40℃反应18~24小时,得到1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐;
(2)合成吡啶基-1,2,3-三唑卡宾钯金属配合物
以乙腈为溶剂,将1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐、氧化银与Me4NCl按摩尔比为1:(1.5~3.5):(5~20)在氮气保护下于20~60℃反应6~8小时,然后加入Pd(CH3CN)2Cl2继续在氮气保护下于20~40℃反应12~24小时,得到吡啶基-1,2,3-三唑卡宾钯金属配合物。
本发明进一步的改进在于,步骤(1)中2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯通过以下过程制得:
以甲苯为溶剂,将7-乙氧羰基吡啶并[1,5-a]四氮唑、苯乙炔、三氟甲磺酸铜按摩尔比为1: (1.5~2):(0.05~0.1)在氮气保护下于80~100℃条件下反应8~12小时,得到2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯。
本发明进一步的改进在于,1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑 -3-鎓四氟硼酸盐与Pd(CH3CN)2Cl2的摩尔比为1:(1.0~1.5);
7-乙氧羰基吡啶并[1,5-a]四氮唑通过以下过程制得:
以甲苯为溶剂,将4-乙氧羰基吡啶氮氧化物、对甲苯磺酰氯、叠氮化钠按摩尔比为1:(2~ 3):(1.5~3)于100~120℃条件下反应18~24小时,得到7-乙氧羰基吡啶并[1,5-a]四氮唑。
一种三唑卡宾钯金属配合物的应用,氮气保护下,向反应容器中加入氯代芳烃、芳基硼酸、碱、三唑卡宾钯金属配合物与反应溶剂,或者向反应容器中加入溴代芳烃、芳基硼酸、碱、三唑卡宾钯金属配合物与反应溶剂,然后在20~120℃下反应2~24小时,反应完成后,经分离纯化得到Suzuki交叉偶联产物。
本发明进一步的改进在于,反应溶剂是乙醇、N,N-二甲基甲酰胺、异丙醇、二氧六环、甲醇与二甲基亚砜中的一种。
本发明进一步的改进在于,碱是碳酸钠、碳酸钾、碳酸铯、氢氧化钠、碳酸氢钾与叔丁醇钾中的一种。
本发明进一步的改进在于,氯代芳烃和碱的物质的量比、溴代芳烃和碱的物质的量比均为1:(1~3),氯代芳烃和芳基硼酸的物质的量比、溴代芳烃和芳基硼酸的物质的量比均为1: (1~2)。
本发明进一步的改进在于,所述的氯代芳烃与三唑卡宾钯金属配合物的物质的量比、溴代芳烃与三唑卡宾钯金属配合物的物质的量比均为1:(0.001~0.1)。
本发明进一步的改进在于,氯代芳烃或者溴代芳烃在反应溶剂中的浓度为0.08~0.16 mol/L;芳基硼酸为苯基硼酸、4-甲基苯基硼酸、4-甲氧基苯基硼酸和3-硝基苯基硼酸中的一种。
本发明与现有技术相比具有以下有益效果:
1、本发明反应条件温和、工艺过程简便,便于操作,设备要求和反应条件容易实现,适宜大规模生产。
2、本发明制备方法简单,催化剂用量小,且催化产率高达98%。
3、本发明在Suzuki偶联反应的应用中,底物适用范围较广、反应混合物经过简单的柱分离,就能高收率的得到高纯度联苯类化合物。
4、实验结果证明,采用本发明三唑卡宾钯金属配合物作为催化剂,Suzuki偶联反应的选择性高、转化率高达100%。
5、通过催化体系的汞毒(Mercury-poisoning)实验对反应机理进行了研究,结果表明三唑卡宾钯金属配合物在催化过程中产生了裸露的零价钯中间体,该中间体可以作为高活性的催化剂诱导Suzuki偶联的顺利进行。
附图说明
下面结合附图对本发明的具体实施方式作进一步详细的说明。
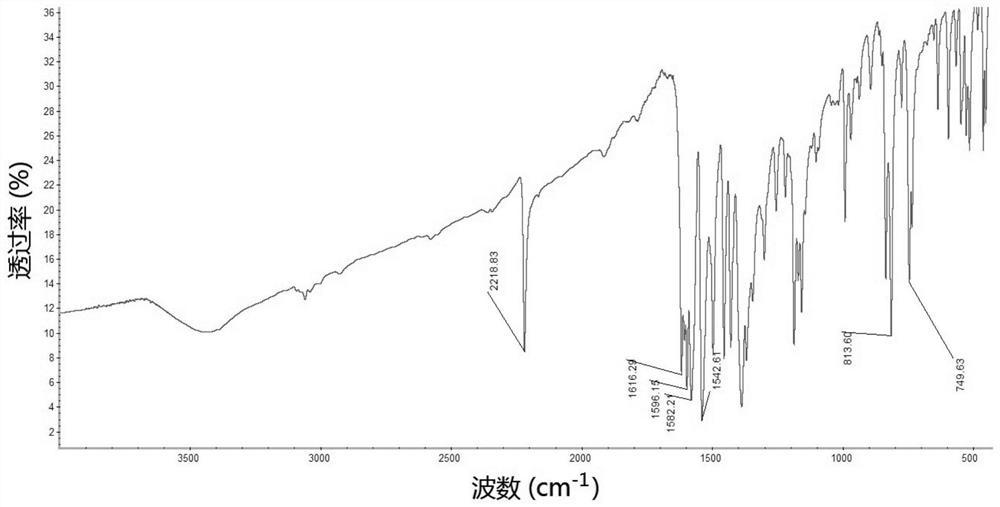
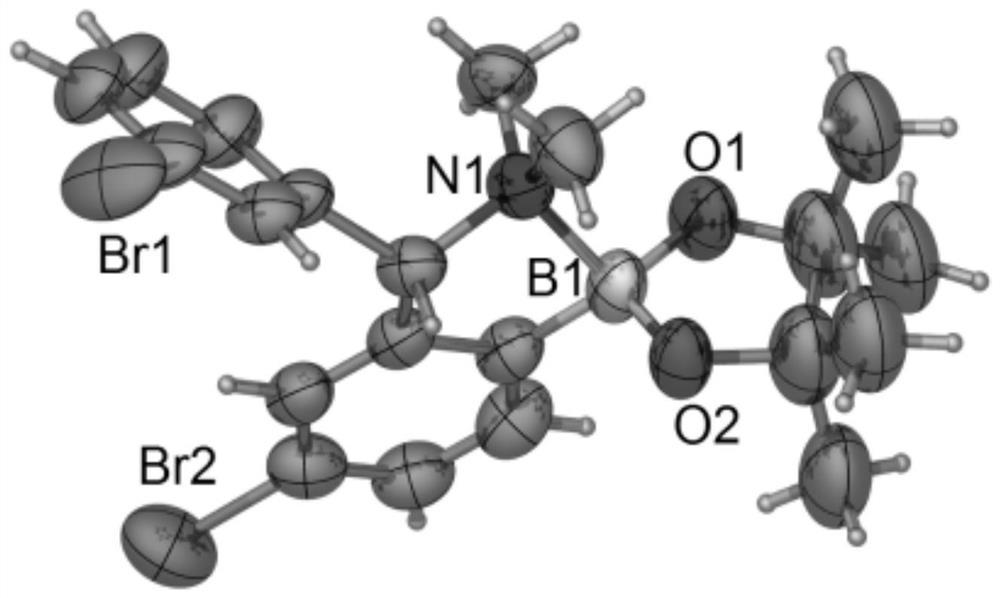
图1为本发明三唑卡宾钯金属配合物单晶谱图。
具体实施方式
为了加深对本发明的理解,下面将结合实施例对本发明做进一步的说明,该实施例仅用于解释本发明,并不构成对本发明的保护范围的限定。实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
本发明的一种三唑卡宾钯金属配合物,以N-吡啶基三唑为分子骨架,以1,2,3-三唑上碳原子和吡啶氮原子与金属钯配位所制备的三唑卡宾钯金属配合物,其化学式为C17H16Cl2N4O2Pd,其结构式为:
如上所述的一种三唑卡宾钯金属配合物的制备方法,包括以下步骤:
(1)合成7-乙氧羰基吡啶并[1,5-a]四氮唑
以甲苯为溶剂,将4-乙氧羰基吡啶氮氧化物、对甲苯磺酰氯、叠氮化钠按摩尔比为1:(2~ 3):(1.5~3)于100~120℃条件下反应18~24小时,反应结束后经柱层析分离纯化,即得到7-乙氧羰基吡啶并[1,5-a]四氮唑;
(2)合成2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯
以甲苯为溶剂,将所述7-乙氧羰基吡啶并[1,5-a]四氮唑、苯乙炔、三氟甲磺酸铜按摩尔比为1:(1.5~2):(0.05~0.1)在氮气保护下于80~100℃条件下反应8~12小时,反应结束后经柱层析分离纯化,即得到2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯;
(3)合成1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐
以二氯甲烷为溶剂,将所述2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯、[Me3O]BF4按摩尔比为1:(1.1~1.5)在氮气保护下于20~40℃反应18~24小时,反应结束后经柱层析分离纯化,即得到1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐;
(4)合成吡啶基-1,2,3-三唑卡宾钯金属配合物
以乙腈为溶剂,将所述1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3- 鎓四氟硼酸盐、氧化银、Me4NCl按摩尔比为1:(1.5~3.5):(5~20)在氮气保护下于20~ 60℃反应6~8小时,然后加入Pd(CH3CN)2Cl2继续在氮气保护下于20~40℃反应12~24 小时,反应结束后经重结晶,即得到吡啶基-1,2,3-三唑卡宾钯金属配合物;
其合成过程如下式:
如上所述的一种三唑卡宾钯金属配合物的晶体结构参见图1,晶体结构的数据见表1,主要键长数据见表2,主要键角数据见表3。
表1三唑卡宾钯金属配合物的晶体学数据
表2三唑卡宾钯金属配合物的主要键长数据
表3三唑卡宾钯金属配合物的主要键角数据
如上所述的一种三唑卡宾钯金属配合物的应用为,该三唑卡宾钯金属配合物作为催化剂在氯代芳烃或者溴代芳烃与芳基硼酸的Suzuki偶联反应中,一步合成联苯类化合物;所述的氯代芳烃与三唑卡宾钯金属配合物的物质的量比、溴代芳烃与三唑卡宾钯金属配合物的物质的量比均为1:(0.05~0.8)。
Suzuki偶联反应合成联苯类化合物的反应通式如下:
具体应用方法为:
氮气保护下,在干燥的Schlenk反应管中依次加入氯代芳烃、芳基硼酸、碱、三唑卡宾钯金属配合物与反应溶剂,或者加入溴代芳烃、芳基硼酸、碱、三唑卡宾钯金属配合物与反应溶剂,然后在20~120℃下反应,反应完成后,得到Suzuki交叉偶联产物。
其中,反应溶剂是乙醇、N,N-二甲基甲酰胺、异丙醇、二氧六环、甲醇与二甲基亚砜中的一种;
碱是碳酸钠、碳酸钾、碳酸铯、氢氧化钠、碳酸氢钾与叔丁醇钾中的一种;
氯代芳烃或者溴代芳烃在反应溶剂中的浓度范围为0.25~0.5mol/L,所述的氯代芳烃和碱的物质的量比、溴代芳烃和碱的物质的量比均为1:(1~3),所述的氯代芳烃和芳基硼酸,溴代芳烃和芳基硼酸的物质的量比均为1:(1~2)。
下面为三唑卡宾钯金属配合物的制备方法的实施例。
实施例1
三唑卡宾钯金属配合物的制备方法,包括以下步骤:
(1)合成7-乙氧羰基吡啶并[1,5-a]四氮唑
分别称量4-乙氧羰基吡啶氮氧化物0.58g(5mmol)、甲苯25mL、对甲苯磺酰氯、叠氮化钠;4-乙氧羰基吡啶氮氧化物与对甲苯磺酰氯的物质的量比为1:2;4-乙氧羰基吡啶氮氧化物与叠氮化钠的物质的量比为1:1.5;将称量好的反应物依次加入带有磁子的100mLSchlenk反应瓶中,室温条件下在磁力搅拌器上搅拌,然后缓慢加热至100℃,用薄层色谱监测反应的进程,24小时后停止反应,冷却至室温,然后直接用柱层析色谱分离,以300~400目的硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离。
所得化合物的物理性质及表征数据如下:
白色固体;
(2)合成2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯
分别称量7-乙氧羰基吡啶并[1,5-a]四氮唑0.95g(4mmol)、甲苯20mL、苯乙炔、三氟甲磺酸铜;7-乙氧羰基吡啶并[1,5-a]四氮唑与苯乙炔的物质的量比为1:1.5;7-乙氧羰基吡啶并[1,5-a]四氮唑与三氟甲磺酸铜的物质的量比为1:0.05;将称量好的反应物依次加入带有磁子的100mL Schlenk反应瓶中,在氮气保护下磁力搅拌器上搅拌,然后缓慢加热至80℃,用薄层色谱监测反应的进程,12小时后停止反应,冷却至室温,用二氯甲烷稀释,用饱和食盐水洗涤三次,有机相用无水硫酸镁干燥,过滤浓缩,用柱层析色谱分离,以300~400目的硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离。
所得化合物的物理性质及表征数据如下:
白色固体;
(3)合成1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐
分别称量2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯1.03g(3.5mmol)和二氯甲烷18mL 依次加入带有磁子的50mL Schlenk反应瓶中,在氮气保护下磁力搅拌器上搅拌,在0℃下用针管滴加[Me3O]BF4,于0℃搅拌5min后缓慢升温至20℃;2-(4-苯基-1H-1,2,3-三唑-1-基) 异烟酸乙酯与[Me3O]BF4的物质的量比为1:1.1;用薄层色谱监测反应的进程,24小时后停止反应,反应液浓缩后经柱层析色谱分离,以300~400目的硅胶作为固定相,以不同比例的甲醇和二氯甲烷的混合溶剂作为洗脱剂对产品进行纯化分离。
所得化合物的物理性质及表征数据如下:
白色固体;
(4)合成三唑卡宾钯金属配合物
分别称量1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐 1.2g(3mmol)、乙腈15mL、氧化银、Me4NCl;1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4- 苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐与氧化银的物质的量比为1:1.5;1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐与Me4NCl的物质的量比为1:5;将称量好的反应物依次加入带有磁子的50mL Schlenk反应瓶中,在氮气保护下于20℃条件下磁力搅拌器上搅拌6小时,然后加入Pd(CH3CN)2Cl2继续在氮气保护下于40℃反应,1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐与Pd(CH3CN)2Cl2的物质的量比为1:1;12小时后停止反应,过滤沉淀,收集、浓缩滤液,用二氯甲烷/正己烷重结晶得到目标产物。
所得化合物的物理性质及表征数据如下:
黄色固体;
实施例2
三唑卡宾钯金属配合物的制备方法,包括以下步骤:
(1)合成7-乙氧羰基吡啶并[1,5-a]四氮唑
分别称量4-乙氧羰基吡啶氮氧化物0.58g(5mmol)、甲苯25mL、对甲苯磺酰氯、叠氮化钠;4-乙氧羰基吡啶氮氧化物与对甲苯磺酰氯的物质的量比为1:3;4-乙氧羰基吡啶氮氧化物与叠氮化钠的物质的量比为1:3;将称量好的反应物依次加入带有磁子的100mLSchlenk 反应瓶中,室温条件下在磁力搅拌器上搅拌,然后缓慢加热至120℃,用薄层色谱监测反应的进程,18小时后停止反应,冷却至室温,然后直接用柱层析色谱分离,以300~400目的硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离。
(2)合成2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯
分别称量7-乙氧羰基吡啶并[1,5-a]四氮唑0.95g(4mmol)、甲苯20mL、苯乙炔、三氟甲磺酸铜;7-乙氧羰基吡啶并[1,5-a]四氮唑与苯乙炔的物质的量比为1:2;7-乙氧羰基吡啶并 [1,5-a]四氮唑与三氟甲磺酸铜的物质的量比为1:0.1;将称量好的反应物依次加入带有磁子的100mL Schlenk反应瓶中,在氮气保护下磁力搅拌器上搅拌,然后缓慢加热至100℃,用薄层色谱监测反应的进程,8小时后停止反应,冷却至室温,用二氯甲烷稀释,用饱和食盐水洗涤三次,有机相用无水硫酸镁干燥,过滤浓缩,用柱层析色谱分离,以300~400目的硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离。
(3)合成1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐
分别称量2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯1.03g(3.5mmol)和二氯甲烷18 mL依次加入带有磁子的50mL Schlenk反应瓶中,在氮气保护下磁力搅拌器上搅拌,在0℃下用针管滴加[Me3O]BF4,于0℃搅拌5min后缓慢升温至25℃;2-(4-苯基-1H-1,2,3-三唑-1- 基)异烟酸乙酯与[Me3O]BF4的物质的量比为1:1.5;用薄层色谱监测反应的进程,24小时后停止反应,反应液浓缩后经柱层析色谱分离,以300~400目的硅胶作为固定相,以不同比例的甲醇和二氯甲烷的混合溶剂作为洗脱剂对产品进行纯化分离。
(4)合成三唑卡宾钯金属配合物
分别称量1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐 1.2g(3mmol)、乙腈15mL、氧化银、Me4NCl;1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4- 苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐与氧化银的物质的量比为1:3.5;1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐与Me4NCl的物质的量比为1:20;将称量好的反应物依次加入带有磁子的50mL Schlenk反应瓶中,在氮气保护下于40℃条件下磁力搅拌器上搅拌8小时,然后加入Pd(CH3CN)2Cl2继续在氮气保护下于20℃反应,1-[4- (乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐与Pd(CH3CN)2Cl2的物质的量比为1:1.5;12小时后停止反应,过滤沉淀,收集、浓缩滤液,用二氯甲烷/正己烷重结晶得到目标产物。
实施例3
(1)合成1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐
以甲苯为溶剂,将4-乙氧羰基吡啶氮氧化物、对甲苯磺酰氯、叠氮化钠按摩尔比为1:2: 2于100℃条件下反应18小时,得到7-乙氧羰基吡啶并[1,5-a]四氮唑。
以甲苯为溶剂,将7-乙氧羰基吡啶并[1,5-a]四氮唑、苯乙炔、三氟甲磺酸铜按摩尔比为1: 1.5:0.1在氮气保护下于100℃条件下反应8小时,得到2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯。
以二氯甲烷为溶剂,将2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯与[Me3O]BF4按摩尔比为1:1.1在氮气保护下于40℃反应18小时,得到1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐;
(2)合成吡啶基-1,2,3-三唑卡宾钯金属配合物
以乙腈为溶剂,将1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐、氧化银与Me4NCl按摩尔比为1:1.5:20在氮气保护下于60℃反应6小时,然后加入Pd(CH3CN)2Cl2继续在氮气保护下于30℃反应12小时,1-[4-(乙氧基羰基)吡啶-2-基] -3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐与Pd(CH3CN)2Cl2的物质的量比为1:1.2;得到吡啶基-1,2,3-三唑卡宾钯金属配合物。
实施例4
(1)合成1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐
以甲苯为溶剂,将4-乙氧羰基吡啶氮氧化物、对甲苯磺酰氯、叠氮化钠按摩尔比为1:3: 1.5于120℃条件下反应24小时,得到7-乙氧羰基吡啶并[1,5-a]四氮唑。
以甲苯为溶剂,将7-乙氧羰基吡啶并[1,5-a]四氮唑、苯乙炔、三氟甲磺酸铜按摩尔比为1:1.7:0.05在氮气保护下于80℃条件下反应12小时,得到2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯。
以甲醇为溶剂,将2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯与[Me3O]BF4按摩尔比为 1:1.5在氮气保护下于30℃反应20小时,得到1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4- 苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐;
(2)合成吡啶基-1,2,3-三唑卡宾钯金属配合物
以乙腈为溶剂,将1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐、氧化银与Me4NCl按摩尔比为1:2.5:10在氮气保护下于40℃反应7小时,然后加入Pd(CH3CN)2Cl2继续在氮气保护下于20℃反应24小时,1-[4-(乙氧基羰基)吡啶-2-基] -3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐与Pd(CH3CN)2Cl2的物质的量比为1:1.3;得到吡啶基-1,2,3-三唑卡宾钯金属配合物。
实施例5
(1)合成1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐
以甲苯为溶剂,将4-乙氧羰基吡啶氮氧化物、对甲苯磺酰氯、叠氮化钠按摩尔比为1: 2.5:3于110℃条件下反应20小时,得到7-乙氧羰基吡啶并[1,5-a]四氮唑。
以甲苯为溶剂,将7-乙氧羰基吡啶并[1,5-a]四氮唑、苯乙炔、三氟甲磺酸铜按摩尔比为1: 2:0.08在氮气保护下于90℃条件下反应10小时,得到2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯。
以二氯甲烷为溶剂,将2-(4-苯基-1H-1,2,3-三唑-1-基)异烟酸乙酯与[Me3O]BF4按摩尔比为1:1.3在氮气保护下于20℃反应24小时,得到1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐;
(2)合成吡啶基-1,2,3-三唑卡宾钯金属配合物
以乙腈为溶剂,将1-[4-(乙氧基羰基)吡啶-2-基]-3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐、氧化银与Me4NCl按摩尔比为1:3.5:5在氮气保护下于20℃反应8小时,然后加入Pd(CH3CN)2Cl2继续在氮气保护下于40℃反应18小时,1-[4-(乙氧基羰基)吡啶-2-基] -3-甲基-4-苯基1H-1,2,3-三唑-3-鎓四氟硼酸盐与Pd(CH3CN)2Cl2的物质的量比为1:1.5;得到吡啶基-1,2,3-三唑卡宾钯金属配合物。
下面为具体的三唑卡宾钯金属配合物的应用的实施例。
实施例6
氮气保护下,在干燥的Schlenk反应管中依次加入间溴甲氧基苯(0.4mmol),芳基硼酸 (0.4mmol),K2CO3(0.4mmol),三唑卡宾钯金属配合物催化剂(0.0004mmol)和DMF(2.5mL);间溴甲氧基苯与芳基硼酸的物质的量比为1:1;间溴甲氧基苯与K2CO3的物质的量比为1:1;间溴甲氧基苯与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.001。然后,将反应管置于20℃的油浴中反应,用薄层色谱监测反应的进程。2小时后,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的Suzuki交叉偶联产物,转化率为94%。
所得化合物的物理性质及表征数据如下:
白色固体;
实施例7
氮气保护下,在干燥的Schlenk反应管中依次加入间溴甲氧基苯(0.4mmol),芳基硼酸 (0.8mmol),Cs2CO3(1.2mmol),三唑卡宾钯金属配合物催化剂(0.04mmol)和EtOH(5mL);间溴甲氧基苯与芳基硼酸的物质的量比为1:2;间溴甲氧基苯与Cs2CO3的物质的量比为1:3;间溴甲氧基苯与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.1。然后,将反应管置于25℃的油浴中反应,用薄层色谱监测反应的进程。3小时后,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的Suzuki交叉偶联产物,转化率高达100%,分离产率高达98%。
实施例8
氮气保护下,在干燥的Schlenk反应管中依次加入间氯甲氧基苯(0.4mmol),芳基硼酸 (0.4mmol),NaOH(0.4mmol),三唑卡宾钯金属配合物催化剂(0.002mmol)和Dioxane(2.5mL);间氯甲氧基苯与芳基硼酸的物质的量比为1:1;间氯甲氧基苯与NaOH的物质的量比为1:1;间氯甲氧基苯与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.005。然后,将反应管置于80℃的油浴中反应,用薄层色谱监测反应的进程。24小时后,将混合物冷却至室温,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的Suzuki交叉偶联产物,转化率为64%。
实施例9
氮气保护下,在干燥的Schlenk反应管中依次加入间氯甲氧基苯(0.4mmol),芳基硼酸 (0.8mmol),Na2CO3(1.2mmol),三唑卡宾钯金属配合物催化剂(0.04mmol)和iPrOH(5mL);间氯甲氧基苯与芳基硼酸的物质的量比为1:2;间氯甲氧基苯与Na2CO3的物质的量比为1:3;间氯甲氧基苯与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.1。然后,将反应管置于120℃的油浴中反应,用薄层色谱监测反应的进程。12小时后,将混合物冷却至室温,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的 Suzuki交叉偶联产物,转化率为86%。
实施例10
氮气保护下,在干燥的Schlenk反应管中依次加入间溴甲基苯(0.4mmol),芳基硼酸(0.4 mmol),tBuOK(0.4mmol),三唑卡宾钯金属配合物催化剂(0.004mmol)和MeOH(3mL);间溴甲基苯与芳基硼酸的物质的量比为1:1;间溴甲基苯与tBuOK的物质的量比为1:1;间溴甲基苯与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.01。然后,将反应管置于 100℃的油浴中反应,用薄层色谱监测反应的进程。16小时后,将混合物冷却至室温,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的Suzuki交叉偶联产物。
所得化合物的物理性质及表征数据如下:
白色固体;
实施例11
氮气保护下,在干燥的Schlenk反应管中依次加入间氯甲氧基苯(0.4mmol),对甲氧基芳基硼酸(0.8mmol),NaHCO3(0.8mmol),三唑卡宾钯金属配合物催化剂(0.04mmol)和DMSO(2.5mL);间氯甲氧基苯与对甲氧基芳基硼酸的物质的量比为1:2;间氯甲氧基苯与NaHCO3的物质的量比为1:2;间氯甲氧基苯与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.1。然后,将反应管置于120℃的油浴中反应,用薄层色谱监测反应的进程。12小时后,将混合物冷却至室温,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的Suzuki交叉偶联产物。
所得化合物的物理性质及表征数据如下:
白色固体;
实施例12
氮气保护下,在干燥的Schlenk反应管中依次加入邻溴甲氧基苯(0.4mmol),芳基硼酸 (0.4mmol),NaHCO3(1.2mmol),三唑卡宾钯金属配合物催化剂(0.004mmol)和DMF(4mL);邻溴甲氧基苯与芳基硼酸的物质的量比为1:1;邻溴甲氧基苯与NaHCO3的物质的量比为1:3;邻溴甲氧基苯与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.01。然后,将反应管置于100℃的油浴中反应,用薄层色谱监测反应的进程。16小时后,将混合物冷却至室温,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的Suzuki交叉偶联产物。
所得化合物的物理性质及表征数据如下:
白色固体;
实施例13
氮气保护下,在干燥的Schlenk反应管中依次加入对溴甲氧基苯(0.4mmol),芳基硼酸 (0.4mmol),tBuOK(0.4mmol),三唑卡宾钯金属配合物催化剂(0.0004mmol)和EtOH(5mL);对溴甲氧基苯与芳基硼酸的物质的量比为1:1;对溴甲氧基苯与tBuOK的物质的量比为1:1;对溴甲氧基苯与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.001。然后,将反应管置于120℃的油浴中反应,用薄层色谱监测反应的进程。12小时后,将混合物冷却至室温,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的Suzuki交叉偶联产物。
所得化合物的物理性质及表征数据如下:
白色固体;
实施例14
氮气保护下,在干燥的Schlenk反应管中依次加入邻溴甲基苯(0.4mmol),芳基硼酸(0.4 mmol),Na2CO3(1.2mmol),三唑卡宾钯金属配合物催化剂(0.004mmol)和Dioxane(2.5mL);邻溴甲基苯与芳基硼酸的物质的量比为1:1;邻溴甲基苯与Na2CO3的物质的量比为1:3;邻溴甲基苯与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.01。然后,将反应管置于 100℃的油浴中反应,用薄层色谱监测反应的进程。16小时后,将混合物冷却至室温,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的Suzuki交叉偶联产物。
所得化合物的物理性质及表征数据如下:
白色固体;
实施例15
氮气保护下,在干燥的Schlenk反应管中依次加入对溴甲基苯(0.4mmol),芳基硼酸(0.4 mmol),NaOH(0.4mmol),三唑卡宾钯金属配合物催化剂(0.04mmol)和iPrOH(5mL);对溴甲基苯与芳基硼酸的物质的量比为1:2;对溴甲基苯与NaOH的物质的量比为1:1;对溴甲基苯与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.1。然后,将反应管置于120℃的油浴中反应,用薄层色谱监测反应的进程。12小时后,将混合物冷却至室温,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的Suzuki交叉偶联产物。
所得化合物的物理性质及表征数据如下:
白色固体;
实施例16
氮气保护下,在干燥的Schlenk反应管中依次加入间溴硝基苯(0.4mmol),芳基硼酸(0.4 mmol),Cs2CO3(1.2mmol),三唑卡宾钯金属配合物催化剂(0.04mmol)和MeOH(2.5mL);间溴硝基苯与芳基硼酸的物质的量比为1:2;间溴硝基苯与Cs2CO3的物质的量比为1:3;间溴硝基苯与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.1。然后,将反应管置于100 ℃的油浴中反应,用薄层色谱监测反应的进程。16小时后,将混合物冷却至室温,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的Suzuki交叉偶联产物。
所得化合物的物理性质及表征数据如下:
白色固体;
实施例17
氮气保护下,在干燥的Schlenk反应管中依次加入2-溴苄腈(0.4mmol),芳基硼酸(0.4 mmol),K2CO3(0.4mmol),三唑卡宾钯金属配合物催化剂(0.04mmol)和DMSO(5mL); 2-溴苄腈与芳基硼酸的物质的量比为1:2;2-溴苄腈与K2CO3的物质的量比为1:1;2-溴苄腈与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.1。然后,将反应管置于120℃的油浴中反应,用薄层色谱监测反应的进程。12小时后,将混合物冷却至室温,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的Suzuki交叉偶联产物。
所得化合物的物理性质及表征数据如下:
白色固体;
实施例18
氮气保护下,在干燥的Schlenk反应管中依次加入1-(4-溴苯基)乙酮(0.4mmol),芳基硼酸(0.8mmol),NaOH(1.2mmol),三唑卡宾钯金属配合物催化剂(0.04mmol)和Dioxane(二氧六环,2.5mL);1-(4-溴苯基)乙酮与芳基硼酸的物质的量比为1:2;1-(4-溴苯基) 乙酮与NaOH的物质的量比为1:3;1-(4-溴苯基)乙酮与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.1。然后,将反应管置于100℃的油浴中反应,用薄层色谱监测反应的进程。16小时后,将混合物冷却至室温,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的Suzuki交叉偶联产物。
所得化合物的物理性质及表征数据如下:
白色固体;
实施例19
氮气保护下,在干燥的Schlenk反应管中依次加入间氯甲氧基苯(0.4mmol),对甲基芳基硼酸(0.4mmol),K2CO3(0.4mmol),三唑卡宾钯金属配合物催化剂(0.004mmol)和iPrOH (5mL);间氯甲氧基苯与对甲基芳基硼酸的物质的量比为1:1;间氯甲氧基苯与K2CO3的物质的量比为1:1;间氯甲氧基苯与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.01。然后,将反应管置于80℃的油浴中反应,用薄层色谱监测反应的进程。24小时后,将混合物冷却至室温,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的Suzuki交叉偶联产物。
所得化合物的物理性质及表征数据如下:
白色固体;
实施例20
氮气保护下,在干燥的Schlenk反应管中依次加入间氯甲氧基苯(0.4mmol),间硝基芳基硼酸(0.8mmol),Na2CO3(1.2mmol),三唑卡宾钯金属配合物催化剂(0.04mmol)和MeOH(4.5mL);间氯甲氧基苯与间硝基芳基硼酸的物质的量比为1:2;间氯甲氧基苯与Na2CO3的物质的量比为1:3;间氯甲氧基苯与三唑卡宾钯金属配合物催化剂的物质的量比为1:0.1。然后,将反应管置于120℃的油浴中反应,用薄层色谱监测反应的进程。12小时后,将混合物冷却至室温,用纯净水稀释,并用乙酸乙酯萃取,合并有机相。有机相依次用稀盐酸溶液和饱和食盐水洗涤,加入无水硫酸钠干燥,过滤浓缩,用柱层析色谱分离,以硅胶作为固定相,以不同比例的乙酸乙酯和石油醚的混合溶剂作为洗脱剂对产品进行纯化分离,得到分析纯的Suzuki交叉偶联产物。
所得化合物的物理性质及表征数据如下:
白色固体;
本发明以N-吡啶基三唑为分子骨架,以1,2,3-三唑上碳原子和吡啶氮原子与金属钯配位所制备的一种三唑卡宾钯金属配合物,其化学式为C17H16Cl2N4O2Pd。本发明还公开了该三唑卡宾钯金属配合物的制备方法及作为催化剂在氯代芳烃或者溴代芳烃与芳基硼酸的Suzuki偶联反应中的应用。本发明具有选择率高、转化率高的特点。
以上所述,仅为本发明的优选实施例,应当指出,对于本技术领域的普通技术人员来讲,在不脱离本发明的核心技术的前提下,还可以做出改进和润饰,这些改进和润饰也应当属于本发明的专利保护范围。与本发明的权利要求书相当的含义和范围内的任何改变,都应认为是包括在权利要求书范围之内的。
一种三唑卡宾钯金属配合物及制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



















动态评分
0.0