



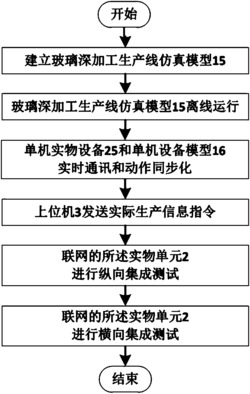



专利摘要
一种番茄红素中间体的制备方法,涉及番茄红素。1)将化合物6溶解在醚类溶剂中,加入对甲苯磺酰肼溶液,再加入化合物7,反应后加入饱和氯化铵淬灭,得化合物8;2)将化合物8溶于乙腈/水溶剂中,加入对甲苯磺酸吡啶盐,反应后用饱和碳酸氢钠溶液淬灭,得粗产物;粗产物溶于苯类溶剂,有机碱以及相同的对甲苯磺酰氯,反应物经饱和氯化铵溶液淬灭、萃取、浓缩、纯化后得化合物9;3)化合物9溶于醚类溶剂中,加入丁基锂试剂,加入单质碘溶液,反应后,经饱和氯化铵溶液淬灭、萃取、浓缩、纯化后得到番茄红素中间体(3E,5E,7E)‑8‑碘‑4‑甲基‑1,3,5,7‑壬四烯。
权利要求
1.一种番茄红素中间体的制备方法,其特征在于包括以下步骤:
1)在0~100℃下,将化合物6溶解在醚类溶剂中,制成溶液A;再加入反应原料2~6倍摩尔数、经有机碱去质子化的对甲苯磺酰肼溶液,使肼与羰基反应以形成苯腙中间体,然后加入与反应原料1~2倍摩尔数的化合物7进行加成反应,反应完全后,加入饱和氯化铵淬灭,经萃取、洗涤、干燥、浓缩、纯化后,得到化合物8;所述化合物6的分子式为C10H18O3,化学名称为由四氢吡喃保护的5-羟基-2-戊酮;所述化合物7的分子式为C8H13OS2,化学名称为3-(2-甲基-1,3-二噻基)丙醛;所述化合物8的分子式为C18H32O3S2,化学名称为(E)-4-甲基-1-(2-甲基-1,3-二噻基)-7-(2H-四氢吡喃-2-氧基)-4-庚-3-烯醇;
2)用步骤1)得到的化合物8制备化合物9,所述化合物9的分子式为C12H18S2,化学名称为2-甲基-2-(4-甲基-2,4,6-庚三烯基)-1,3-二噻烷,具体制备方法如下:
(1)将化合物8溶于乙腈/水溶剂中,制备成溶液B,然后加入反应原料0.1~1倍摩尔数的对甲苯磺酸吡啶盐,并在30~100℃加热搅拌,促使发生THP脱保护形成二醇,反应结束后冷却并用饱和碳酸氢钠溶液淬灭,经萃取,浓缩后得粗产物;
(2)将步骤(1)得到的粗产物溶于苯类溶剂,加入反应原料1~5倍摩尔数的有机碱以及对甲苯磺酰氯,并在30~100℃下加热搅拌,反应完全后,反应物经饱和氯化铵溶液淬灭、萃取、洗涤、干燥、浓缩、纯化后,得化合物9;
3)在惰性氛围以及-100~50℃条件下,将步骤2)得到的化合物9溶于醚类溶剂中,制成溶液C,然后加入反应原料1~3倍摩尔数的丁基锂试剂,在惰性氛围下加入反应原料2~4倍摩尔数的单质碘溶液,反应完全后,经饱和氯化铵溶液淬灭、萃取、洗涤、干燥、浓缩、纯化后得到番茄红素中间体(3E,5E,7E)-8-碘-4-甲基-1,3,5,7-壬四烯。
2.如权利要求1所述一种番茄红素中间体的制备方法,其特征在于在步骤1)中,所述醚类溶剂选自四氢呋喃或乙醚。
3.如权利要求1所述一种番茄红素中间体的制备方法,其特征在于在步骤1)中,所述溶液A的摩尔浓度为0.5~10mol/L。
4.如权利要求1所述一种番茄红素中间体的制备方法,其特征在于在步骤1)中,所述对甲苯磺酰肼的用量是化合物6摩尔数的1~3倍;所述有机碱的用量是化合物6摩尔数的2~6倍,反应物的摩尔浓度是0.5~10mol/L;所述有机碱选自甲醇钠、乙醇钠、叔丁醇钾中的一种,所述化合物7是化合物6摩尔数的1~2倍。
5.如权利要求1所述一种番茄红素中间体的制备方法,其特征在于在步骤1)中,所述萃取,选用乙酸乙酯萃取;所述洗涤,选用饱和氯化钠洗涤;所述干燥,选用无水硫酸钠或无水硫酸镁。
6.如权利要求1所述一种番茄红素中间体的制备方法,其特征在于在步骤2)中,所述化合物8的摩尔浓度是0.5~5mol/L;所述溶液B的摩尔浓度为0.5~5mol/L;所述对甲苯磺酸吡啶盐当量是反应物的摩尔数为0.1~1.0倍;所述有机碱是反应物的1~5倍,所述有机碱选自DBU、三乙胺、吗啡啉中的一种;所述乙腈与水的体积比是1/2~3/1。
7.如权利要求1所述一种番茄红素中间体的制备方法,其特征在于在步骤2)中,所述苯类溶剂选自苯、甲苯、二甲苯中的一种;所述萃取,选用乙酸乙酯萃取;所述洗涤,选用饱和氯化钠洗涤;所述干燥,选用无水硫酸钠或无水硫酸镁。
8.如权利要求1所述一种番茄红素中间体的制备方法,其特征在于在步骤3)中,所述化合物9的摩尔浓度是0.1~10mol/L;所述丁基锂的用量是化合物9摩尔数的1~3倍;所述碘用量是化合物9摩尔数的2~4倍;所述氯化铵浓度选用饱和。
9.如权利要求1所述一种番茄红素中间体的制备方法,其特征在于在步骤3)中,所述醚类溶剂选自四氢呋喃或乙醚,所述单质碘溶液溶解于干燥的甲苯中,I2的摩尔浓度为0.5~2mol/L。
10.如权利要求1所述一种番茄红素中间体的制备方法,其特征在于在步骤3)中,所述萃取,选用二氯甲烷萃取;所述洗涤,选用饱和氯化钠洗涤;所述干燥,选用无水硫酸钠或无水硫酸镁。
说明书
技术领域
本发明涉及番茄红素,尤其是涉及一种番茄红素中间体的制备方法。
背景技术
作为类胡萝卜素中的一员,番茄红素的化学式为C40H56,以下称化合物1,结构式如下:
由于番茄红素具有高效除单线态氧自由基的能力,因此被广泛应用于防治衰老、心脏疾病以及癌症等方面;它还可以作为一种优良的食品添加剂,用于生产各类保健品,因此番茄红素的需求量与日俱增。由于番茄红素在天然产物中含量不高,天然提取的番茄红素远远无法满足市场日益增长的需要。化学合成法制备番茄红素具有产量大、成本低廉以及快速高效的优点,因此成为各国医药企业以及研究人员的研究热点。
传统的合成工艺主要采用Wittig方法(Hoffmann-La Roche Inc.Method for the manufacture ofcarotinoids and novel intermediates:US,5166445[P].1990-01-11.;BASF Aktiengesellschaft.Preraration of phosphonium salts:US,6187959B1[P].1998-08-07);近期Angel R.de Lera等人(Chem.Commun.,2013,49,2694-2696)报道了一种采用有机硼试剂对烯基碘进行贵金属钯催化偶联的番茄红素高效合成路线,该方法路线简洁高效,可适用于多种类胡萝卜素的合成,也不需要用到昂贵且毒性较大的三苯基膦,因此具有很好的工业化应用前景。其合成路径如下:
其中使用了对称的双烯基碘化合物——(2E,4E,6E,8E,10E,12E,14E)-2,15-二碘-6,11-二甲基-2,4,6,8,10,12,14-庚烯(分子式C20H26I2,以下简称化合物2)与有机硼试剂——2-((1E,3E)-4,8-二甲基-1,3,7-丙烯)-4,4,5,5-四甲基-1,3,2-二氧环戊硼烷(分子式C17H29O2B,以下简称化合物3)进行钯催化偶联反应得到番茄红素。而化合物2可以由两分子的化合物4经烯烃复分解反应一步得到。其中化合物3可以方便地由廉价易得的天然产物香叶醇经两步反应制备而成,因此合成番茄红素的关键在于化合物4的制备。Angel R.de Lera等(B.Dom1′nguez,B.Iglesias and A.R.de Lera,Tetrahedron,1999,55,15071-15098)在制备化合物4的过程中用到剧毒而又昂贵的有机锡试剂——(E)-乙基-3-三丁基锡烷基-2-丁烯酸(以下简称化合物5),且步骤烦琐,产率偏低,这既增加了反应的成本,也不利于环境保护,难以实现工业化生产。
发明内容
本发明所要解决的技术问题是克服上述现有化合物4的合成技术存在的合成步骤长、总收率低的缺点,提供原料廉价易得、合成路线简洁可行的一种番茄红素中间体的制备方法。
本发明采用的技术路线为:
所述一种番茄红素中间体的制备方法包括以下步骤:
1)在0~100℃下,将化合物6溶解在醚类溶剂中,制成溶液A;再加入反应原料2~6倍摩尔数、经有机碱去质子化的对甲苯磺酰肼溶液,使肼与羰基反应以形成苯腙中间体,该苯腙中间体在过量的有机碱作用下发生Bamford-Steven反应形成热力学稳定的烯基负离子,然后加入与反应原料1~2倍摩尔数的化合物7进行加成反应,反应完全后,加入饱和氯化铵淬灭,经萃取、洗涤、干燥、浓缩、纯化后,得到化合物8;所述化合物6的分子式为C10H18O3,化学名称为由四氢吡喃保护的5-羟基-2-戊酮;所述化合物7的分子式为C8H13OS2,化学名称为3-(2-甲基-1,3-二噻基)丙醛;所述化合物8的分子式为C18H32O3S2,化学名称为(E)-4-甲基-1-(2-甲基-1,3-二噻基)-7-(2H-四氢吡喃-2-氧基)-4-庚-3-烯醇;
2)用步骤1)得到的化合物8制备化合物9,所述化合物9的分子式为C12H18S2,化学名称为2-甲基-2-(4-甲基-2,4,6-庚三烯基)-1,3-二噻烷,具体制备方法如下:
(1)将化合物8溶于乙腈/水溶剂中,制备成溶液B,然后加入反应原料0.1~1倍摩尔数的对甲苯磺酸吡啶盐,并在30~100℃加热搅拌,促使发生THP脱保护形成二醇,反应结束后冷却并用饱和碳酸氢钠溶液淬灭,经萃取,浓缩后得粗产物;
(2)将步骤(1)得到的粗产物溶于苯类溶剂,加入反应原料1~5倍摩尔数的有机碱以及相同的对甲苯磺酰氯(TsCl),并在30~100℃下加热搅拌,反应完全后,反应物经饱和氯化铵溶液淬灭、萃取、洗涤、干燥、浓缩、纯化后,得化合物9;
3)在惰性氛围以及-100~50℃条件下,将步骤2)得到的化合物9溶于醚类溶剂中,制成溶液C,然后加入反应原料1~3倍摩尔数的丁基锂试剂,使化合物9的硫醚键充分开环形成硫鎓中间体(类似反应的文献详见Chemical Reviews,2000,100(8)3187-3204),之后在惰性氛围下加入反应原料2~4倍摩尔数的单质碘溶液,反应完全后,经饱和氯化铵溶液淬灭、萃取、洗涤、干燥、浓缩、纯化后得到番茄红素中间体(3E,5E,7E)-8-碘-4-甲基-1,3,5,7-壬四烯,即化合物4。
在步骤1)中,所述化合物6可由商品化的5-羟基-2-戊酮和3,4-二氢吡喃在DMF的作用下,经60℃反应8h一步制备而成,详细步骤见Synthetic Communications,1995,25(13):1981-1987;所述醚类溶剂可选自四氢呋喃或乙醚等;所述溶液A的摩尔浓度可为0.5~10mol/L;所述化合物7可由1,3-二噻烷在ZnEt2催化下经一锅法合成,制备方法详见Tetrahedron:Asymmetry,2000,11(5):1067-1071。所述对甲苯磺酰肼的用量最好是摩尔数的1~3倍;所述有机碱的用量最好是摩尔数的2~6倍,反应物的摩尔浓度最好是0.5~10mol/L;所述有机碱可选自甲醇钠、乙醇钠、叔丁醇钾等中的一种,所述化合物7最好是摩尔数的1~2倍;所述萃取,可选用乙酸乙酯萃取;所述洗涤,可选用饱和氯化钠洗涤;所述干燥,可选用无水硫酸钠或无水硫酸镁等。
在步骤2)中,所述化合物8的摩尔浓度最好是0.5~5mol/L;所述溶液B的摩尔浓度可为0.5~5mol/L;所述对甲苯磺酸吡啶盐当量最好是反应物的摩尔数为0.1~1.0倍;所述有机碱最好是反应物的1~5倍,所述有机碱可选自DBU、三乙胺、吗啡啉等中的一种;所述乙腈与水的体积比最好是1/2~3/1。所述苯类溶剂可选自苯、甲苯、二甲苯等中的一种;所述萃取,可选用乙酸乙酯萃取;所述洗涤,可选用饱和氯化钠洗涤;所述干燥,可选用无水硫酸钠或无水硫酸镁等。
在步骤3)中,所述化合物9的摩尔浓度最好是0.1~10mol/L;所述丁基锂的用量最好是摩尔数的1~3倍;所述碘用量最好是摩尔数的2~4倍;所述氯化铵浓度可选用饱和;所述醚类溶剂可选自四氢呋喃或乙醚等,所述单质碘溶液可溶解于干燥的甲苯中,I2的摩尔浓度可为0.5~2mol/L。所述萃取,可选用二氯甲烷萃取;所述洗涤,可选用饱和氯化钠洗涤;所述干燥,可选用无水硫酸钠或无水硫酸镁等。
本发明的原料来源方便,路线简单可行,成本低廉,可以用于工业化生产。制备的番茄红素中间体也同样可作为其它类胡萝卜素的合成原料,因此具有很好的应用价值和发展潜力。
具体实施方式
下面通过实施例对本发明做进一步的说明。
实施例1
(1)将对甲苯磺酰肼(12.2g,100mmol)溶于四氢呋喃溶液(500mL)中,制成0.4mol/L溶液,再加入甲醇钠(16.2g,300mmol)。反应物在室温搅拌1h后,降至0℃,然后缓慢加入化合物6(18.6g,100mmol)的THF(10mL)溶液继续搅拌5h,然后缓慢添加化合7(28.5g,150mmol),升至30℃搅拌12h后加入饱和氯化铵淬灭,减压除去有机溶剂,加水稀释后用乙酸乙酯萃取(150mL×3),合并有机相,用饱和氯化钠溶液洗涤,无水硫酸钠干燥。经过滤、浓缩、纯化得到30.4g无色液体化合物8(产率约为85%)。
产品结构确认:IR(film)νmax:3216,2917,1480,1441,1402,1259,1026,761cm-1;1H NMR(400MHz,DMSO-d6)δ5.73(s,1H),5.41(dt,J=1.5Hz,7.8Hz,1H),4.58(t,1H,J=8.0Hz),4.18(t,J=7.8Hz,1H),3.74-3.64(m,2H),3.41(t,J=6.9Hz,2H),2.47-2.37(m,4H),2.28-2.199(m,2H),2.13-2.03(m,4H),1.98-1.84(m,6H),1.76-1.43(m,8H);δ139.7,122.0,106.4,77.9,69.5,65.7,63.3,30.5,29.7,28.7,28.5,26.2(2C),25.8,25.4,21.9,20.4,12.5;MS(ESI)m/z 363(M+Na+,100%);HRMS-ESI Calcd for C18H32S2O3(M+H+):361.1871;found:361.1865.
(2)步骤一:将化合物8(18g,50mmol)与对甲苯磺酸吡啶盐(960mg,5mmol)溶解于乙腈/水溶液(500mL,v/v=1/1)中,70℃加热搅拌3h反应结束后用200mL饱和碳酸氢钠淬灭,分离有机相,水相用乙酸乙酯萃取(50mL×3),合并有机相,用饱和氯化钠溶液洗涤,无水硫酸钠干燥。经过滤、浓缩后直接作为原料投入下步骤二反应;
步骤二:将步骤一中的粗产物溶解于甲苯溶液(500mL),加入20.8mL DBU(19.6g,150mmol)搅拌30min后加入对甲苯磺酰氯(9.45g,50mmol)的甲苯溶液(30mL),在80℃下搅拌12h,反应结束后加入饱和氯化铵淬灭,水相用乙酸乙酯萃取,合并有机相,先用无水硫酸钠干燥。有机相经浓缩、纯化得到9.67g目标产物,即化合物9,产率81%,产品为黄色液体。
产品结构确认:IR(film)νmax:2913,2848,1659,1424,1347,1293,955,764,658cm-1;1H-NMR(400MHz,C6D6):6.55(d,J=10.8Hz,1H),6.48-6.39(m,1H),6.18-6.06(m,1H),6.03(d,J=14.8Hz,1H),5.30(d,J=16.4Hz,1H),5.08(d,J=10.0Hz,1H),2.60(d,J=14,4Hz,2H),2.47-2.37(m,4H),2.16(s,3H),2.13-2.01(m,2H),1.64(s,3H)ppm 13C NMR(100MHz,CDCl3)δ171.9,169.2,167.5,144.9,134.7,134.1,133.9,132.1,130.0,129.9,129.6,129.2,1290,126.6,124.2,53.7,29.7,27.4,15.3;MS(ESI)m/z 221(M+H+,100%);HRMS-ESI Calcd for C13H20S2(M+H+):241.1085;found:241.1098.
(3)在氮气保护以及-78℃下,将化合物9(2.4g,10mmol)溶于四氢呋喃溶剂(50mL)中,之后缓慢加入丁基锂溶液(4mL,20mmol),在该温度搅拌30min,接着缓慢加入单质I2(3.05g,12mmol)的四氢呋喃溶液(10mL)至反应完全。然后加入100mL饱和氯化钠淬灭,水相用乙酸乙酯萃取,合并有机相,先用无水硫酸钠干燥。有机相经浓缩、纯化得到1.82g目标产物,即化合物3,产率70%,产品为黄色液体。
产品结构确认:IR(film)νmax:2913,2848,1659,1424,959cm-1;1H-NMR(400MHz,C6D6):6.84(d,J=10.8Hz,1H),6.63-6.53(m,1H),6.15-6.04(m,2H),5.98(d,J=15.2Hz,1H),5.20(d,J=16.8Hz,1H),5.08(d,J=10.1Hz,1H),2.18(s,3H,CH3),1.61(s,3H)ppm.13C-NMR(100.62MHz,C6D6):δ141.5(d),138.0(d),135.3,133.7,133.5,123.4,118.4,97.0,28.0,12.4;MS(ESI)m/z 261(M+H+,100%);HRMS-ESI Calcd for C10H14I(M+H+):261.0140;found:261.0148.
本发明以廉价易得的化合物6为原料,首先在有机碱作用下发生Bamford-Steven反应后与化合物7加成得到化合物8,接着在质子酸的作用下发生THP水解得到二醇中间体,然后在碱性条件下与对甲苯磺酰氯反应并消除得到化合物9;最后在丁基锂的作用下,化合物9经开环后与单质碘加成并发生消除反应得到目标产物——一种番茄红素中间体,即化合物4;各步骤操作以及分离简单,路线较短,产率较高,试剂均为常用廉价试剂,适合大量生产。
一种番茄红素中间体的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。


















动态评分
0.0